


- 232.75 KB
- 10页

- 1、本文档共5页,可阅读全部内容。
- 2、本文档内容版权归属内容提供方,所产生的收益全部归内容提供方所有。如果您对本文有版权争议,可选择认领,认领后既往收益都归您。
- 3、本文档由用户上传,本站不保证质量和数量令人满意,可能有诸多瑕疵,付费之前,请仔细先通过免费阅读内容等途径辨别内容交易风险。如存在严重挂羊头卖狗肉之情形,可联系本站下载客服投诉处理。
- 文档侵权举报电话:19940600175。
'甾体药物结构改造的一般方法甾体药物的基本碳架具有一个环戊烷骈多氢菲的母核和三个侧链。“甾”字形象地表示了该类化合物的结构特征,即表示在含有四个稠合环“田”字上面连有三个侧链“巛”。一般说来其中两个是角甲基,另一个为含不同碳原子数的碳链。不管是通过半合成还是全合成方法合成甾体药物,都必须对甾体母核各个特定部位进行化学结构改造和修饰,才能使药物和受体有效地进行定向结合,才能发挥药理作用。本次培训主要对甾体化合物结构改造的一般方法按A、B、C、D四环的顺序,作一简要总结,旨在与各位同事互相交流和讨论,共同进步。一.A环的结构改造:㈠C1-C2上的结构改造:在甾体母核A环C1-C2处引入双键,是皮质激素的基本结构之一,如醋酸泼尼松、地塞米松、倍他米松等,常用的有以下两种方法:1.微生物法:利用节杆菌产生的脱氢酶将C1-C2单键转变为双键,这是目前各类皮质激素普遍采用的主导方法。本法收率较高,成本较低,但生产周期较长。2.化学法:肤轻松的合成可采用SeO2进行1,2位脱氢,如:㈡C3上的结构改造:C3碳原子是甾体化合物最活泼的部位,能和许多化学试剂发生反应,人们充分利用这一特点对甾体化合物进行了大量的结构改造,合成了许多具有生物活性的化合物。1.C3上常见的基本集团为:
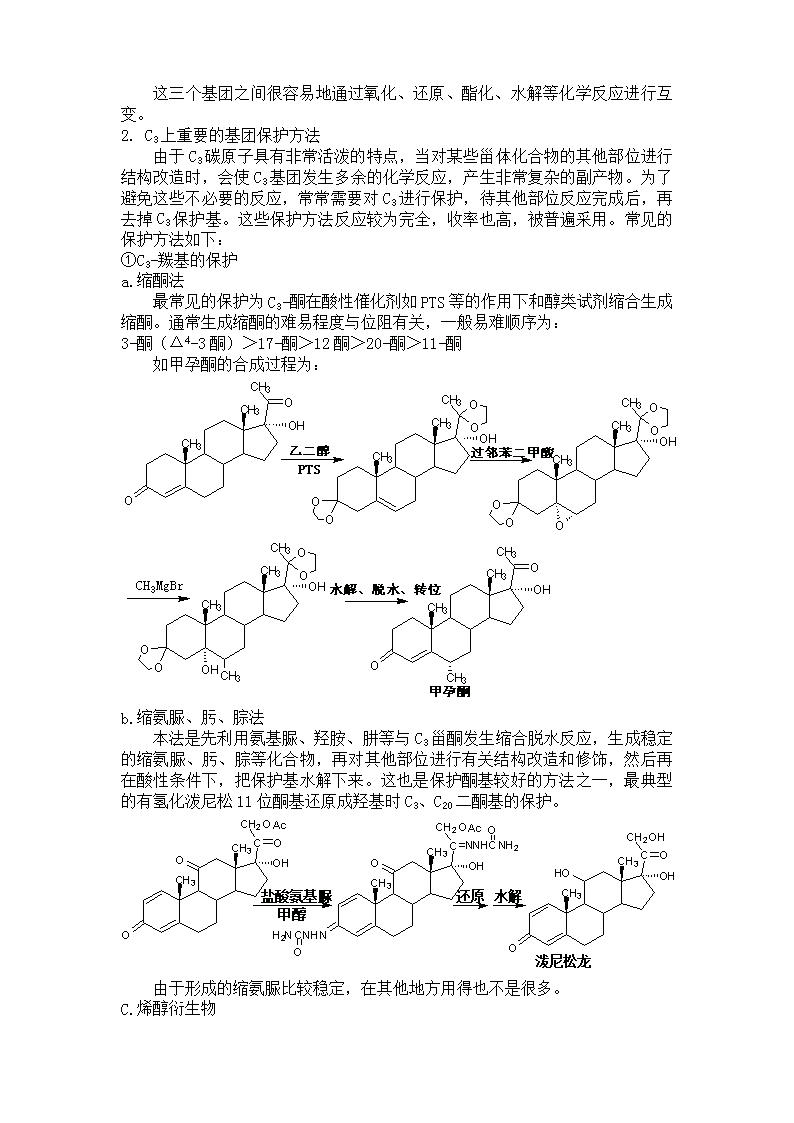
这三个基团之间很容易地通过氧化、还原、酯化、水解等化学反应进行互变。2.C3上重要的基团保护方法由于C3碳原子具有非常活泼的特点,当对某些甾体化合物的其他部位进行结构改造时,会使C3基团发生多余的化学反应,产生非常复杂的副产物。为了避免这些不必要的反应,常常需要对C3进行保护,待其他部位反应完成后,再去掉C3保护基。这些保护方法反应较为完全,收率也高,被普遍采用。常见的保护方法如下:①C3-羰基的保护a.缩酮法最常见的保护为C3-酮在酸性催化剂如PTS等的作用下和醇类试剂缩合生成缩酮。通常生成缩酮的难易程度与位阻有关,一般易难顺序为:3-酮(Δ4-3酮)>17-酮>12酮>20-酮>11-酮如甲孕酮的合成过程为:b.缩氨脲、肟、腙法本法是先利用氨基脲、羟胺、肼等与C3甾酮发生缩合脱水反应,生成稳定的缩氨脲、肟、腙等化合物,再对其他部位进行有关结构改造和修饰,然后再在酸性条件下,把保护基水解下来。这也是保护酮基较好的方法之一,最典型的有氢化泼尼松11位酮基还原成羟基时C3、C20二酮基的保护。由于形成的缩氨脲比较稳定,在其他地方用得也不是很多。C.烯醇衍生物
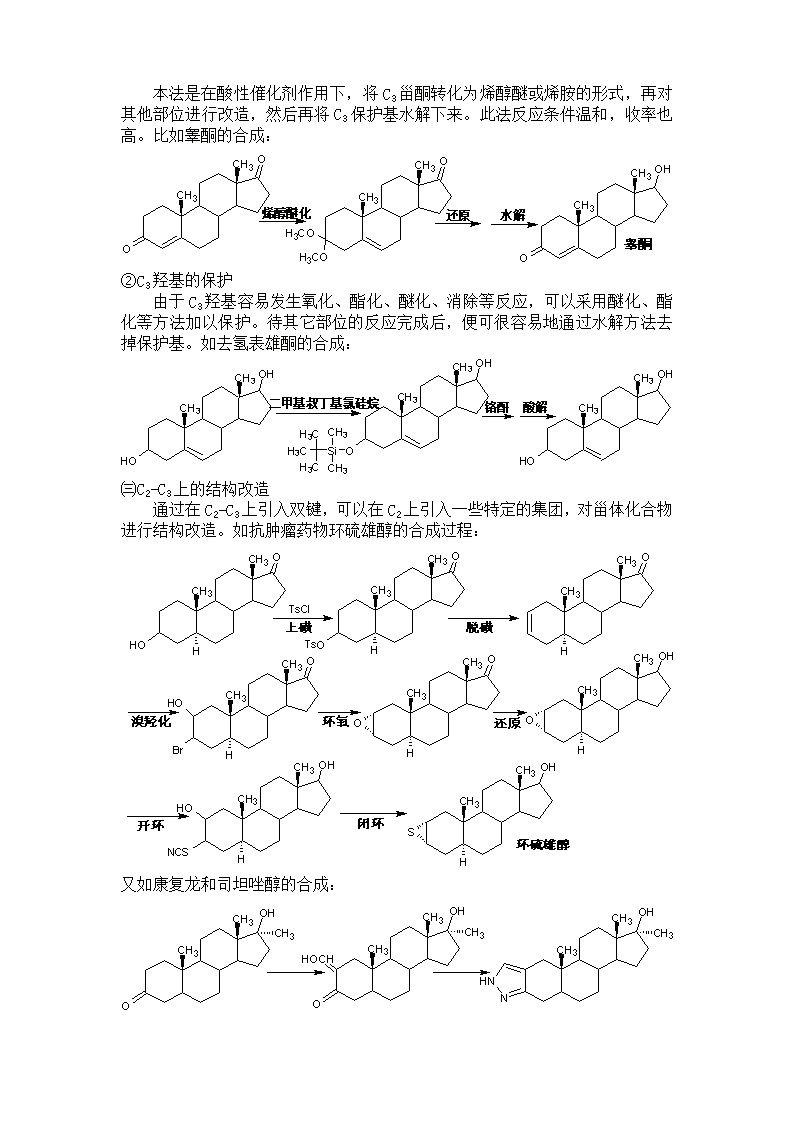
本法是在酸性催化剂作用下,将C3甾酮转化为烯醇醚或烯胺的形式,再对其他部位进行改造,然后再将C3保护基水解下来。此法反应条件温和,收率也高。比如睾酮的合成:②C3羟基的保护由于C3羟基容易发生氧化、酯化、醚化、消除等反应,可以采用醚化、酯化等方法加以保护。待其它部位的反应完成后,便可很容易地通过水解方法去掉保护基。如去氢表雄酮的合成:㈢C2-C3上的结构改造通过在C2-C3上引入双键,可以在C2上引入一些特定的集团,对甾体化合物进行结构改造。如抗肿瘤药物环硫雄醇的合成过程:又如康复龙和司坦唑醇的合成:
㈣A环芳构化将A环芳构化,再经过一系列结构改造,可以合成许多雌性激素。如炔诺酮中间体开环物经节杆菌脱氢酶脱氢芳构化即可得到雌酚酮,由雌酚酮可合成许多雌性激素如雌二醇、雌三醇及其衍生物等。㈤A环形成杂环结构在A环引入杂原子可得到一些具有特定生理活性,能调节男性机体内分泌系统的药物。通过将Δ4双键氧化,再引入胺基,再在C1,2位、C17位进行相应的改造,可得到对5α-还原酶具有较好抑制作用的药物,如非那雄胺等。非那雄胺对男性前列腺增生、前列腺癌、男性脱发具有明显疗效。合成过程如下:㈥A环形成降(Nor)或高(Homo)碳化合物。如双炔失碳酯的合成:二.B环结构的改造B环的结构改造主要是通过在C6、C7上引入一些特定的基团;或者通过在C5-C10-C9-C11碳链上形成亲核电子链,以便在相应部位引入特定基团;或者将C6上的基团与C19上的基团进行环合、开环,在C19上引入醇羟基,再经氧化、脱羧去掉C19碳原子,从而得到一些具有特定生物活性的化合物。1.在C6上引入甲基
如甲羟孕酮、6α-甲基强的松龙的合成过程中,C6甲基的引入主要有以下两种方法:①先以过酸形成5,6环氧,再进行格氏反应、脱水、转位制得甲羟孕酮:②也可先进行17酯化,再经醚化、迈尼希反应上C6次甲基后,经加氢转位制得甲羟孕酮:2.C6上引入卤素,C7上引入特定基团通过在C5,C6之间引入双键和环氧,可以在C6上引入卤素。如氯地孕酮的合成过程:
又如肤轻松的合成过程:又如安体舒通的合成:3.通过在C5-C10-C9-C11碳链上引入亲核电子链,可在C11上引入相应的基团。如米非司酮的合成:
4.通过将C6上的基团与C19上的基团进行环合、开环,在C19上引入醇羟基,再经氧化、脱羧去掉C19碳原子,从而形成蛋白同化激素的关键中间体:三.C环上的结构改造1.C11-羟基的引入在C11位引入羟基有生物发酵和化学合成两种方式,化学合成工艺由于生产成本较高在工业化生产中通常不予采用;利用犁头霉、黑根酶、新月弯胞霉等真菌胞内酶的氧化作用引入C11-羟基仍然是合成各种皮质激素最基础、最经济的传统方式。引入的羟基可以经过普氏氧化转变成酮基,酮基又可以在硼氢化钾的作用下变成具有药理活性的C11β羟基。2.C9-氟的引入利用C11-羟基可在C9、C11之间引入双键,经溴羟化加成反应、环氧化、氟化氢加成在C9上引入氟原子,这是合成含氟皮质激素的最常用方法。
3.C11上引入其他基团如米非司酮的合成过程:4.C12-酮基的改造在甾体资源—番麻皂素的利用上,需要对C12碳原子进行改造。四.D环上的结构改造1.C20、C21碳链的去除通过贝克曼重排将C20、C21碳原子去掉,可合成各种性激素及相关激素。2.C17碳原子上的结构改造①C17酮基的还原:如睾酮的合成过程
②C17-羟基的酯化③C17酮基的加成3.C16、C17碳原子上的结构改造C16、C17碳原子上的结构改造常常通过先形成C16、17的α环氧化合物,再通过加成、氧化、酰化、醚化等反应接上其他基团,用以合成目标化合物。①C17碳原子上引入羟基②在C16、C17上同时引入集团:如格氏反应、开环反应等
如16α羟基泼尼松龙的合成又如阿孕奈德关键中间体—双羟孕酮的合成①C21碳原子上的结构改造C21碳原子上的氢原子比较活泼,可以发生卤素取代反应,再进行置换反应引入其他基团。如氢化可的松中间体RSA的合成;再在C21羟基上接上不同基团来调节甾体药物的脂溶性和水溶性,如地塞米松磷酸钠等。'
您可能关注的文档
- 建设工程结构改造实施流程方案
- 浅谈非标60 mw供热汽轮机组汽封环及油挡结构改造
- 车立方钢结构改造工程施工组织设计
- 建设工程结构改造实施流程方
- 某承重结构改造指导书
- 老旧小区结构改造工程屋面工程装饰装修工程水暖工程电气工程维修改造施工组织设计
- 结构改造工程技术标
- 兖矿鲁南化肥厂原料及动力结构改造项目造粒机电梯设备采购招标文件
- 910d型减速器壳体应力分析与结构改造
- 2016年度路网结构改造安全防护工程招标文件标书.doc
- 住宅建设工程结构改造实施流程方案
- 建设工程结构改造实施流程方案
- 单层及多层厂房的结构改造设计
- 2013年公路路网结构改造工程(地质灾害)监理计划
- 住宅建设工程结构改造实施流程方案
- 建设工程结构改造实施流程方案
- 路面结构改造工程施工组织设计
- 单层及多层厂房的结构改造设计.doc