


- 1.43 MB
- 89页

- 1、本文档共5页,可阅读全部内容。
- 2、本文档内容版权归属内容提供方,所产生的收益全部归内容提供方所有。如果您对本文有版权争议,可选择认领,认领后既往收益都归您。
- 3、本文档由用户上传,本站不保证质量和数量令人满意,可能有诸多瑕疵,付费之前,请仔细先通过免费阅读内容等途径辨别内容交易风险。如存在严重挂羊头卖狗肉之情形,可联系本站下载客服投诉处理。
- 文档侵权举报电话:19940600175。
'天津大学硕士生论文AbstractOpticalactivecompound1waspreparedviaMannichreactionfromL-prolineand2-naphth01.whichWasthenesterifiedwithSOCL,andMeOHtogivelIinhighyield.Grignardreactionof11withseveralGrignardreagentsaffordedaseriesofopticallyactivetridentateaminoalcobolligandsIV—VⅡI.ThescligandswerecomplexcdwitllLAHscpcratelyandthenusedtopromotetheasynunetricMichaeladditionofdiethylmalonateto2-cyclohexenoneorchalcone.Thechemicalyieldswereexcellent.Unfortunately,theopticalyieldswerelow.I,II,Ⅳ一ⅥIIarenewcompouds.111eirstructureswereidentifidedbyelementalanalysis、IRand1HNM眠.AseriesofsinomeninederivativesweresynthesizedviaGrignardreaction.n地structureofderivativeXⅡWasdeterminedbyelementalanalysis、Ⅱt、1HNM噼and”CNMRandtestifiedwithX-rayanalysis.ThoroughidentificationofothercompoundsⅪH.-xⅨiSongoing.SinomeninewasreducedwithNaB地andtllestructureoftheproductWBSidenfifidedbyelementalanalysis、IR、1HNMR、”CNMRandX-rayanalysis.Excellentregeo-andstereo—selectivitywereobservedinboth也ereductionand例gnardreaction.Diaminescondencedwellwithsinoarninedione。A11theabovederivativeswillbeteStedfortheirpossiblepainreleasinge丑’ect.Keywords:tridentatearninoalcoholligandsL-prolineMichaelAdditionReactionsinomenineanddione
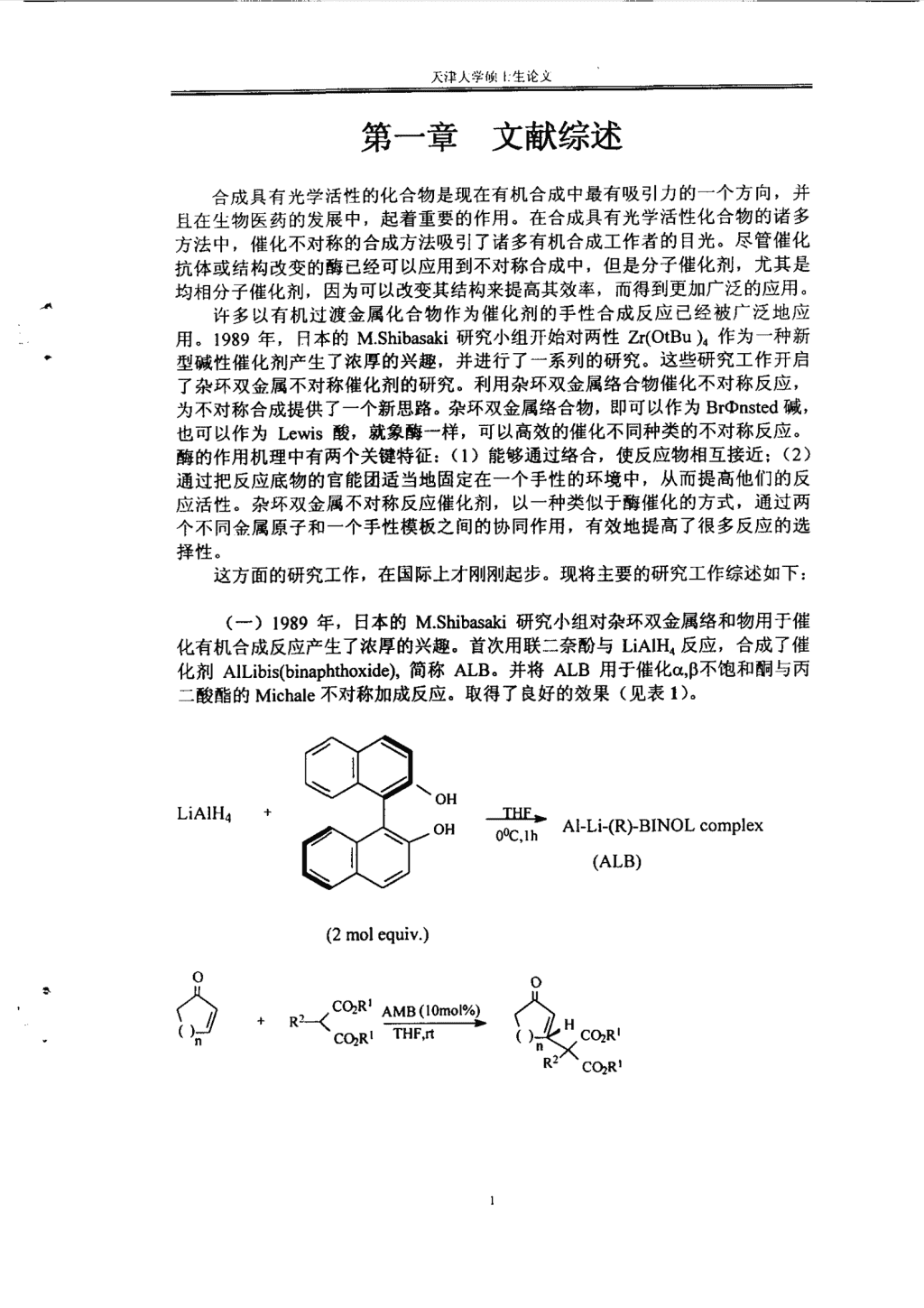
——————————————————————————————————一——一———————————————————————————————————————————————————————一第一章文献综述合成具有光学活性的化合物是现在有机合成中最有吸引力的一个方向,并且在生物医药的发展中,起着重要的作用。在合成具有光学活性化合物的诸多方法中,催化不对称的合成方法吸引了诸多有机合成工作者的目光。尽管催化抗体或结构改变的酶已经可以应用到不对称合成中,但是分子催化剂,尤其是均相分子催化剂,因为可以改变其结构来提高其效率,而得到更加广泛的应用。许多以有机过渡金属化合物作为催化剂的手性合成反应已经被广泛地应用。1989年,日本的M.Shibasaki研究小组开始对两性Zr(OtBu)。作为一种新型碱性催化剂产生了浓厚的兴趣,并进行了一系列的研究。这些研究工作开启了杂环双金属不对称催化剂的研究。利用杂环双金属络合物催化不对称反应,为不对称合成提供了一个新思路。杂环双金属络合物,即可以作为Brmnsted碱,也可以作为Lewis酸,就象酶一样,可以高效的催化不同种类的不对称反应。酶的作用机理中有两个关键特征:(1)能够通过络合,使反应物相互接近;(2)通过把反应底物的官能团适当地固定在一个手性的环境中,从而提高他们的反应活性。杂环双金属不对称反应催化剂,以一种类似于酶催化的方式,通过两个不同会属原子和~个手性模板之间的协同作用,有效地提高了很多反应的选择性。这方面的研究工作,在国际上才刚刚起步。现将主要的研究工作综述如下:(一)1989年,日本的M.Shibasaki研究小组对杂环双金属络和物用于催化有机合成反应产生了浓厚的兴趣。首次用联=奈酚与LiAIH。反应,合成了催化剂AILibis(binaphthoxide),简称ALB。并将ALB用于催化0‘,B不饱和酮与丙二酸酯的Michale不对称加成反应。取得了良好的效果(见表1)。(2molequiv.)OH。H苌等Al-Li_(胁BINOLc。mplex(ALB)OC02R≯薰l眦A一荟.蚕.<酽。弋∥。/.、(
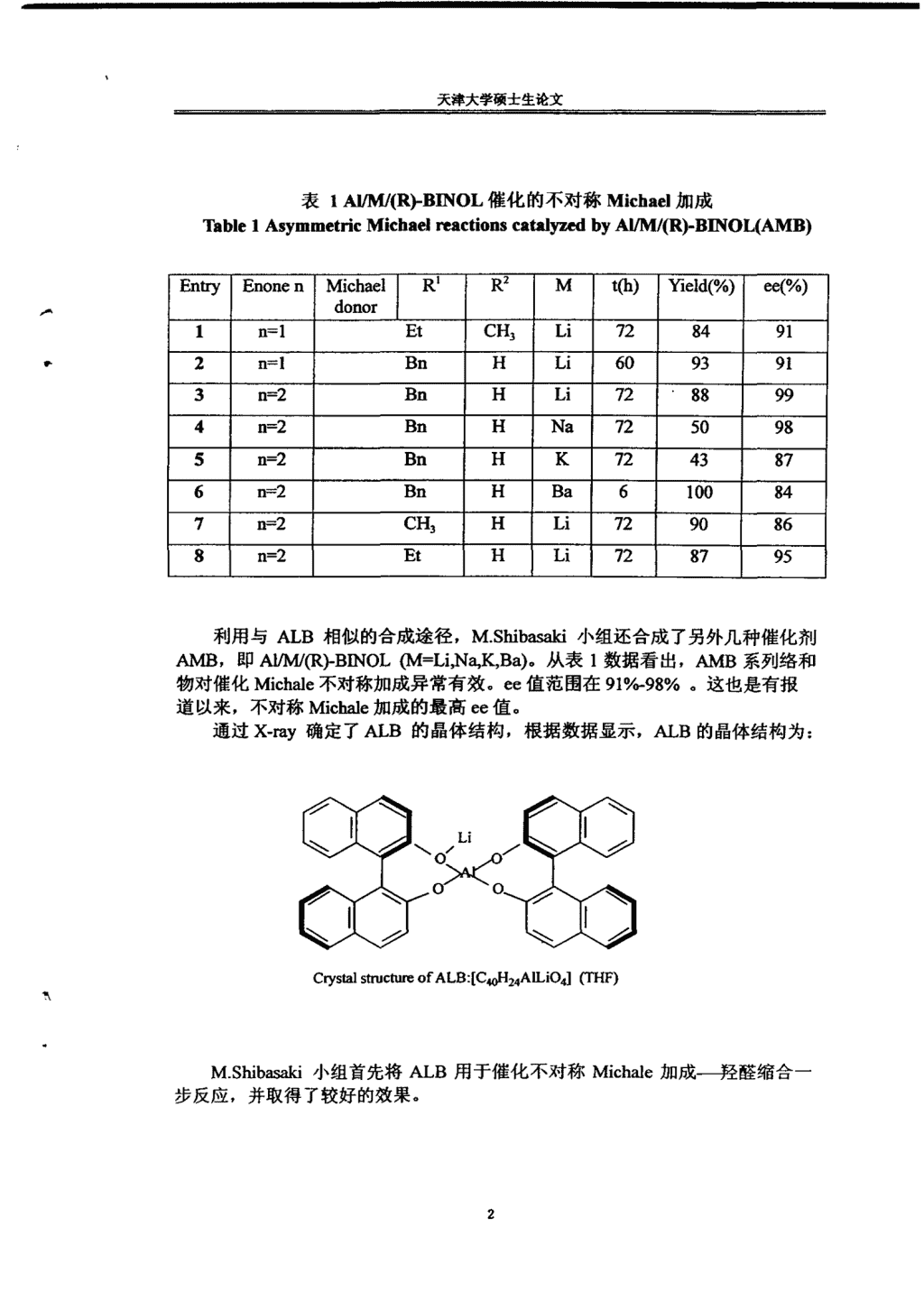
天津大学硕士生论文表1AI/M/(R)-BINOL催化的不对称Michael加成Table1AsymmetricMichaelreactionscatalyzedbyAI/M/(R)-BINOL(AMB)EntryEnonenMichaelR1R2Mt(h)Yield(%)ee(%)donor1n=lEtCH3Li7284912n=lBnHLi6093913n=2BnHLi7288994n=2BnHNa7250985n=2BnHK7243876n=2BnHBa6100847n=2CH3HLi7290868n=2EtHLi728795利用与ALB相似的合成途径,M.Shibasald小组还合成了另外几种催化剂AMB,即AI/M/(R)-BINOL(M=Li,Na,K,Ba)。从表l数据看出,AMB系列络和物对催化Miehale不对称加成异常有效。ee值范围在910/o-98%。这也是有报道以来,不对称Michale加成的最高ee值。通过X-my确定了ALB的晶体结构,根据数据显示,ALB的晶体结构为:CwstalstructureofALB:【C.oH24AlLi04】frrtF)M.Shibasaki小组首先将ALB用于催化不对称Michale加成——羟醛缩合一步反应,并取得了较好的效果。
天津大学硬士生论文凸+c“嚣+一cm警oC02Et+C02Et7Yield(*/,)90删64Yield(%)9lee(%)当用苯甲醛代替氢化肉桂醛反应时,得到两种异构体组成的混合物,用吡啶氯代铬酸盐(简称PCC)进一步氧化得到相应的二酮衍生物。产物为一种立体异构体(89%ee)。pO+c吣=+Dcno卫}82Yield(%)(diastvneomixture)lOOYield(%)89ee(%)一步催化的Miehfle—aldol反应历程可描述如下:甲基丙二酸酯首先与ALB络合生成相应的锂烯醇盐1,1进攻已与舢络合的环戊酮的双键,得Al烯醇盐ll,ll进一步和醛反应,得到醇盐111。111上的OH基的质子H被取代,生成三组分反应产物,并重新生成了ALB。从而完成了催化反应循环(见Figure11。3
重量奎耋至圭竺篁兰m◆oR三FigureI因工M,Shibasaki研究小组成功地将这一机制用于合成11--deoxy-PGF。6采‘s’ALB(10”1%、86哌91%∞)4cooc}bEtOH1L—d∞xy—PGF
天津大学硕士生论文为了进一步验证ALB的催化性能,M.SMbasaki研究小组将ALB应用于醛的磷酰化反应。ORCHO+出ocH3)2坠!竺!!型兰!THF..●oOC得到生理活性的a_羟基膦酸。而后者可作为中间体,制备一系列具有重要生理作用的a-羟基取代的磷酰基化合物。实验证明,在合成a-羟基膦酸中,ALB有良好的催化性能(见表2)。表2ALB催化的醛的不对称磷酰化反应。Table2AsymmetrichydrophosphonylationofaldehydescatalyzedbyALBEntryAldehyde--Rm)Yield(%)ee(%)lPh5195902—N02C扎40857l3p_℃lC6H,3880834p_CH3C池9282865p-CH30C6H11588786p-(CH3hNCpq,一【a】7(E)-PhCH=CH83828(E)-PhCH--CH(CH3)6180569(E卜CH3(CH2)2CH=CH39855510C也(cH93CH24l9516从表中数据可以看出,醛上有拉电子取代基时,产率和∞值有较大的提高。Al是第1II主族元素,鉴于此,M.Shibasaki小组以B,Ga.orIn为中心正离子合成了一些杂环双金属催化剂。C,-a/Na/BINOL(binaphthoxide){简称GaSB)和In/K/BINOL{简称ZnPB}能够有效地催化环己酮和丙二酸酯的Michale不对称加成反应。但是B/alkalimental/BINOL络和物对上述反应没有作用(见表3)。<篡AM2"O★【毒w/、vo,"k5OC02Bn挽∞HjO叫^。◇
表3杂环双金属催化剂(10m01%)催化的环己烯酮与丙二酸酯的不对称Michael加成反应Table3AsymmetricMichaelreactionof2-cyclohexen-I—onewithmalonatecatalyzedbyaheterobimetallicAsymmetriccatalyst(10m01%)EntryM1M2t01)Yield[%]eel%】lBLi17O2BNa21O3BK1504G-aLi437l495G-aNa14345986GaK4450867InLi247728InNa9525109hK1686184结果表明,GaSB的对映选择性较InPB好。GaSB的合成见下式:CrysmlstructureofGaSB6oN8型!塑,ONart,lhG:◇★
天津大学硕士生论文室温下,10m01%GaSB催化的环己酮与丙二酸酯的不对称Michale反应需要143h。尽管产率为45%,ee%值为98。但是时间过长。M.Shibasaki小组尝试着加入添加剂,以缩短反应时间。以10m01%与9m01%的丁醇钠的混合物作为催化剂,催化a,B不饱和酮与丙二酸二苯酯的不对称加成反应,21h得到98%ee%值和87%的收率。1989年,M.Shibasaki小组合成了两性化合物Zr(OtBu),。并将其用于羟醛缩合反应。反应式如下。但这些羟醛缩合反应都是在化学计量的Zr(OtBu).催化下进行的。当采用催化计量Zr(OtBu)4时,结果不尽如意。OZr(OtBu)4+~肋等oOH§+—一嘶尝764%,100%basedOnrecovered2-cyclopentenonezI(Oteu)4(2.5molequiv)TI-IF,-300c56*/0forml鼬打p。ll反水+B一蚓竺坚呱患
;;圣童奎兰至圭兰墼三Zr(O旧uhf2.OmolEluivTI-F.-3盯℃·rtoAldolreaction、vitllZr(OtBu)4;THP=tetrahydropyranyl,MS=methanefonyl,DBU=I,8-diazabicyclo[5.4.0]undee-7-eneG.shib嬲姑小组认为原因在于ⅣB族元素的醇盐,蔗性不够。并设想如果采用稀土元素的醇盐作为催化剂效果比较好。原因是稀土元素离解能(5.4_6.4ev)★电负性(1.1一l_3)都比较低。PhCH0+Oc认O....r.a.r—e..e.a.r—t—h——a—l—k—o——x—i—d—e—s-.TI-IFPhoHOLa3(OtBu)9(3.3m01%),--720C,74%Y3(OtBu)sClf3.3m01%),一430C,50%燃。粤净OHcH3NO:≤凳}价人,N02明:1而:i虿+吖\/~M2{rareeart}halkoxides嘲+硝詈蹋+踊abLa3(OtBu)9(3.3m01%),a40%,b30%Y5(OtPr)130(3.3m01%),a100%随后,G.Shibaski研究小组以稀土元素为核心元素,开展了一系列研究,合成了LnMB(Ln_稀土元素,M—碱金属,B---Binaphthoxide联二奈酚)。并将其用于8
天津大学硕士生论文合成重要的医药中间体及具有重要生理功能的化合物。LLB(La/Ln/BINOL)的合成路线如下(见figure2):LaCl3·7H20+La(OtPr)34-(3molequiv.)Figure2NaotBuLLBcomplexthen}120(1molequiv.)后经x-测定,LLB(La/Ln/BINOL)的晶体结构为:
天津大学硕士生论文为了检验LLB的催化性能,G.Shib鹊h研究小组将其用于催化不对称硝基羟醛反应。∥丫删。NPIll0H+CH3N02{鬻鬻7屿2hP吖coon哪’_40℃,一-⋯一NPtll取得了令人满意的结果(见表4)。表4LLB催化的非对应选择性硝基羟醛反应Table4DiastereoselectivenitroaldolreactioncatalyzedbyLLBEntryCatalystYield[%]Erythro:threoEe(etythro)[%】1(R)-LLB9299:1962(S)-LLB9674:2690M.shibaski小组推测硝基羟醛缩合反应的机理如下:RCHO醛的磷酰化反应,在LLB的催化下,结果令人满意。实验数据证明LLB,是一种优良高效的不对称催化剂。
R删。+潭(OCH3):翌TH筹F8。驾C少F弛k,—7oln表5LLB催化的醛的不对称磷酰化反应Table5AsymmetrichydrophosphonylationofaldehydescatalyzedbyLLBEntryR“h)Yield[%]ee%1Ph888792o-N02C丑1285363p-CIC脚.880634r,-CHlCd-I-793785I>CH;OC,I-h98388[a16o-CROC。}{I887937p-(CH,),NC。H.128895fal8p-(CH,)NC。H.1280959(E)一PhCH=CH8908410(E)-PhCH=C(CH,)8949211(E)一CH,(CH,),CH=CH8637512CH,(C如),CR88861反应机理如下:RCHoLSB(L=La,S=Na)作为一种不对称催化剂,对于硝基羟醛反应效果不理想,但对于催化Michael加成反应,却有着良好的性能
.,COORlcat.+R2_85%从以上数据看出,杂环双金属络和物2对Michael加成反应非常有效。丙二酸酯与0【,B不饱和酮.醛.硝基化合物,在2催化下,反应得到较高的收率而且反应时间逐渐缩短。加成产物的对映体过量比已报道的文献值要好。总之,杂环双金属化合物,以类似于酶催化的方式,有效的催化很多种类的不对称加成反应。这些催化剂一方面起着路易斯酸的作用,另一方面起着布郎苔德碱的作用。简言之,杂环双金属化合物,作为手性配体催化剂,已经成功地用于不对称加成反应。取得了令人满意的结果,已经尝试着用于工业生产。并且为不对称合成开创了新的方法,思路。
天津大学硕士生论文第二章实验部分日本的M.Shibasaki及印度的G.Manickam、G.Sundarajan研究小组制备的杂环双金属络合物,作为不对称反应催化剂,活性很高,产物收率和ee值的结果令人满意。但催化剂所采用的原料较难制备。因此我们采用有手性中心的氨基酸与B一萘酚通过曼尼期(Mannichreaction)反应缩合在一起。合成一系列配体。反应路线如下:第一节实验路线1手性氨基醇的合成路线:1.1B.奈酚和脯氨酸,在甲醇溶液中,与甲醛反应,反应式如下:◎了伽+锒c删华C∞H0HI1.21甲酯化。甲醇做溶剂,I先和二氯亚砜反应,然后和甲醇发生甲酯化反应,反应式如下:c028里醛。OHSOCl2IⅡC02CH3OHc=OIo曲蹬凹蹬∞譬的
天津大学硕士生论文1.3II在无水.无氧条件下,与一系列Gringard试剂反应。合成一系列手性氨基醇,反应式如下:PhBr+Mg等PhMgBrC止Igar+Mg亡焉≯C4H9MgBrc,2蛐+Mg丢焉≯C12H25MgBrc2H5Br+Mg丢篙≯C2H5MgBrIIMg亡焉≯+RMgBr』竿2催化的AsymmetricMichael加成反应:2.1chaleone的制备,反应式如下:C695CHO+COHsCOCH3—塑虬C6H5CH=CHCOC6H52.22.3.环己酮的制备,反应式如下:l8一螂叫》譬∞
垂兰奎耋塑圭兰丝兰Br2+oc芍L一嘶犷丑00H20/CH3CH20H\/L、夕2.3Michael不对称加成反应反应一反应二(5-c腻n-℃o+LiAlIl4+cKCcOQ2EEtt/一DO⋯⋯嗽蒜一文畋嚣Iv(未鉴定)Vl9VI(未鉴定)
重童查兰矍圭篁丝三VIII(未鉴定)第二节实验过程1试剂来源,规格,及各种测量仪器:1.1试剂规格及来源试剂名称规格2-奈酚ARL-prolineAR二氯亚砜AR镁屑四氢呋哺乙酸乙酯三乙胺甲醛溴苯溴乙烷氯乙酰氯GR5149.85对甲基苯磺酰氯溴代异丙烷溴代十二烷环己酮乙醚石油醚氢化铝锂溴素碳酸钠氯仿无水硫酸钠乙醇苯乙酮苯甲醛亮氨酸苯酰氯ARCRARCPARCP生产厂家中国医药公司北京采购供应站经销ACROSORGANICS(进口1天津市化学试剂一厂中国新奥化学开发公司武汉天津市化学试剂一厂天津市东天正精细化学试剂厂天津化学试剂二厂天津市化学试剂批发公司中国常州市光明生物化学研究所北京亚太精细化工公司中国余山化工厂上海中国上海试剂一厂上海北京理工大学宏宇化工公司北京市化学试剂公司天津市东天正精细化学试剂厂德国天津市试剂一厂天津市化学试剂二厂天津市化学试剂一厂天津市化学试齐J一厂天津市东天正精细化学试剂厂天津市化学试剂~厂北京市化学试剂公司ACROSORGANICS(进口)上海试剂一厂2O
天津大学硕士生论文丁二酰氯硅胶254柱色谱硅胶青藤碱氯苄DM噼DMSO乙二胺邻苯二胺碳酸胍1.2实验仪器:名称电光天平旋转薄膜蒸发仪定时恒温旋转搅拌器熔点测定仪红外光谱仪核磁共振仪旋光仪元素分析仪2溶剂的精制:CP上海试剂一厂200-260目青岛海洋化工厂200-260目青岛海洋化工厂CP中国医药公司北京采购供应站经销AR天津市大港一中化工厂AR天津津东天正精细化学试剂厂AR天津市试剂一厂AR天津津字精细化学试剂厂AR北方芳草医药化工研制公司CP军事医学科学院药材供应站型号D7-100ZFQ-5.型规格或生产厂家0-99.999g天津玻璃仪器厂上海虹浦仪器厂Ⅵ州AGIMOTO~IP.500BIO-BADEXAI,IBURFT$3000BRI,KERAC20024lMCPerk.in-ElmerYANACoCHNCORERⅣ匝.3精制乙醚的方法:在少量二苯酮存在下,乙醚与Na回流(N2保护)至溶液变为紫黑色,然后蒸出即可。"H-IF的精制的方法:在少量二苯酮存在下,哪与Na回流,至溶液变为紫黑色,然后蒸出即可。若回流时间过长,可以先将TI-IF蒸出,在加入Na,在二苯酮存在下回流,这样可以大大缩短时间。整个过程都需要在N2保护下。无水溴乙烷的制备:称量C2HsBr100ml,加入无氧化二磷3_49,回流2.3h,然后蒸出即可,无水保存备用。无水溴苯的制各:同无水溴乙烷的制备。甲醇的精制:甲醇50ml倒入500ml圆底烧瓶中,加入59镁屑和0.29碘。加热回流,使镁屑全部溶解后,再加入0.19碘,使反应加速(但总量(O.39)。镁全溶后,加入100ml甲醇,回流lh后,蒸馏,无水保存。无水溴代十二烷的制备:同无水溴乙烷的制备。无水溴代异丙烷的制备:同无水溴乙烷的制备。无水溴丁烷的制备:同无水溴乙烷的制备。无水氯苄的制各:同无水溴乙烷的制备。3手性氨基醇的合成:21
MW271.3(1)称量一定量的2.奈酚(2-Naphthol,MW144.2,分子式CloH80,2.6579,18.4mml01)和脯氨酸(L-Proline,MW115.1,分子式C5t-Ic-q022.1229,18.4ret001)溶于■醇中,搅拌.(2)滴加甲醛20ml,滴加完毕后,搅拌反应3.5ll,停止反应。反应过程中,TLC跟踪检测反应进程。减压蒸馏除去多余甲醛。蒸去溶剂。(TLC检测一个点)冷冻,结晶,过滤。滤饼抽干。得产物4.5179,产率90.4%。红外,1HNMR谱图显示得到了产物。元素分析结果:1分子式为Cl。Hl,N03:C%70.81(70.85):H%6.31(6.27);N%5.16(5.17)。取删,趴。:。曲IoH⋯85。3曲6⋯s。3(1)称量1(MW271.3,4.5179,16.6ret001)溶于无水甲醇(35m1),搅拌,冰盐浴冷却(-5‘C)。(2)不断搅拌下,滴加二氯亚砜(3ml,MW)。反应物由不溶转而溶解。滴加完毕后,搅拌0.5ll,加热回流。反应液由无色,逐渐加深,至橙红色。反应4.5h,停止反应。(3)向反应体系中,加入饱和NaCO,溶液,出现白色固体。调中性(PH=7)。乙酸乙酯萃取(30mlX3),合并有机相(TLc检测一个点,氯仿:三乙胺_2:0.2)。无水NaSO,干燥有机相,除去乙酸乙酯。反应液结晶,过滤,真空干燥。得块状晶体2.8959。产率82%,mpl26.127。红外,一HNMR谱图显示得到了产物II。元素分析结果:Ⅱ分子式为Cl,H。9NO,:C%71.57(70.58);H%6.70(6.67);N%4.89(4.91)。IVMw409.32噼侧蛩叼
天津大学硕士生论文(1)溴苯格氏试剂的制备:称量一定量的镁屑(MW24.1,1.8569,76.4ret001),加入精制的乙醚(40m1),并加入一粒碘,用于引发格氏反应。称量无水溴苯(MWl57.分子式ce%Br,11.9889,76.4ret001),溶于无水乙醚(20m1),加入到恒压滴液漏斗中。抽真空,充N:保护。引发反应(微热)后,搅拌下,滴加溴苯和无水乙醚配成的溶液。调节滴加速度,使反应保持迅速回流。滴加完毕后,加热回流2h。(2)称量II(MW285.3,29),加入到反应体系中。反应液颜色加深,由黄色变为亮黄色,并有沉淀生成。室温搅拌0.5h后(TLC检测仍有原料,氯仿:石油醚_2:1),加热回流反应2.5h。停止反应,加氯化铵溶液调中性。用乙醚(30ml×3)萃取,合并后,用无水NaSoI干燥有机相,除去溶剂。真空干燥,得产品IV3.2609,产率94.192%。冷冻保存待用。VMW369.5(1)溴丁烷格氏试剂的制备:称量一定量的镁屑(MW24.1,6.789,278.9ret001),加入加入精制的THF(70m1),加入沸石,一粒碘(用于引发格氏反应)。称量无水溴丁烷(MWl37,分子式C.‘I-IgBr,38.1909,278.9ret001),无水I-IF(50m1),加入到恒压滴液漏斗中。抽真空,充N:保护。加热引发反应后,搅拌下,滴加溴丁烷和无水TIIT配成的溶液。调节滴加速度,使反应保持稳定的回流。滴加完毕后,加热回流2h。将制好的溴丁烷格氏试剂无水无氧储存备用。(2)称量II(MW285.3,0.59),溶于10ml无水n忭,搅拌溶解。用冰浴冷却至5。C,N:保护下,加入C4HoMgBr(MWl61.4,2.6229)。反应}陵由浅红色变为浅黄色。搅拌反应12h,TLC检测反应液,原料消失,氯仿展开。停止反应。反应液中加入冰水,氯化铵溶液调中性。用乙醚(20ml×3)萃取,合并后,用无水NaSq干燥有机相,除去溶剂。真空干燥,得产品610mg,产率92.7%。红外,HNMR谱图显示得到了产物V.冷冻保存待用。VIMw593.5(1)溴代正十二烷格氏试剂的制备:方法同溴丁烷格氏试剂的制备。制好后,无水无氧储存备用。(2)称量II(MW285.3,O.59),溶于10ml无水THF,搅拌溶解。冰浴冷却至(<5‘C)。N2保护下,加入c12H25MgBr(MW275.3,3.8359)。反应液由浅红色变为亮黄色。TLC跟踪检测反应进程(氯仿:三乙胺--2=O.2)。23
天津大学硕士生论文搅拌反应5。TLC检测,原料消失。停止反应。后处理(同V)。产品用TLC检查一个点。冷冻保存待用。MW313.4(1)溴乙烷格氏试剂的制备:方法同溴丁烷格氏试剂的制备。制好后,无水无氧储存备用。(2)称量Ⅱ(MW285.3,0.29),溶于5ml无水四氢呋喃,搅拌溶解。冰浴冷却至(<5。C)。抽真空,N2保护下,加入CqHsMgBr(MWl33.3,748mg)。反应液由浅红色变为亮黄色。TLC跟踪检测反应进程(氯仿:三乙胺=2:O.2)。拌反应3.5h。反应完毕,后处理(同V)。TLC检测一个点。蒸去溶剂,真空干燥,得产品187,产率85.1%。红外,HNMR谱图显示得到了产物VII。冷冻保存待用.MW341.4(1)异溴丙烷格氏试剂的制备:方法同溴丁烷格氏试剂的制备。制好后,无水无氧储存备用。(2)称量II(MW285.3,0.29),溶于5ml无水四氢呋喃,搅拌溶解。冰浴冷却至(.(5‘C)。抽真空,N2保护下,加入C3HTMgBr(MWl47.3,826mg)。反应液由浅红色变为亮黄色。TLC跟踪检测反应进程(氯仿:三乙胺=2:0,2)。拌反应6.5h。反应完毕,后处理(同Ⅵ。TLC检测一个点。蒸去溶剂,真空干燥,得产品VIIl216mg,产率90.0%。冷冻保存待用。亚苄基苯乙酮(chalchon)的制备:将129NaOH与98ml水配成的溶液加入到500ml厚壁烧杯中,冰浴冷却下,加入509乙醇。在激烈搅拌下,向溶液中加入269(0.22m01)苯乙酮,再加入239(0.24m01)苯甲醛。将反应物在15.300C搅拌2.3h。反应物逐渐稠化,以至于搅拌困难。停止反应。冷冻,过滤,滤饼用水洗涤至中性。后用乙醇(10ml,00C)洗涤。粗品重结晶(乙醇中),得浅黄色晶体产品389,产率84%。24
天津大学硕士生论文2,3一环己烯酮的制备:(1)溴化:称量一定量的环己酮(Mw98.1,分子式cdql00),溶于255ml甲醇中。室温搅拌下,滴加少量的Br2。待颜色褪去,引发反应后。冰浴冷却,控制温度15.20。C,滴加剩余的溴素(MW160,分子式Br2,32.6409,204ret001)。(要求反应液保持淡黄色,Br’滴加完毕时,反应液的浅黄色要保持数分钟,否则补加Br2)。反应液倾倒至51gNa2CO,和204ml石油醚的混合液中。搅拌0.5h后,加入255ml水。过滤除去不溶物。转移至分液漏斗中,静置分层。收集有机相。水层用石油醚(50ml×1)萃取,合并有机相。无水Na2S04干燥有机相,蒸去溶剂。剩余物减压蒸馏。得产物43.2899(820C,6mmHg)。产率95.2%。(2)合成2,3一环己烯酮:称量第一步的产物(MW223.1,18.29),加入甲醇95.4ml,NaOH(MW40.0,23.99)。加热回流反应3天,TLC检测(乙醚:石油醚=l:2),反应完毕。将反应液倾入到160ml饱和食盐水中,过滤。石油醚(30mlx3)萃取。有机相用无水硫酸钠干燥。除去石油醚。向得到的产品中,加入等体积3%稀硫酸,振荡5分钟。用乙醚(30mix3)萃取。合并有机相,用稀溶液洗涤。干燥后,除去乙醚。剩余油状物,减压蒸馏,的产品(MW96.1,5.2889),产率67.5%。4Michael不对称加成反应:4.1催化计量的V用于charlton的Michael加成:称量V(MW369.5,109mg,0.3mm01),溶于无水THF(Sml)中。抽真空,充N"气保护下,加入LiAlI-h(MW37.5,23mg,0.6ret001)。冰浴冷却下,搅拌0.5h。称量亚苄基苯乙酮(MW208.3,208mg,lmm01),丙二酸二乙酯(MW160.2,192mg,1.2mm01),加入反应体系中。室温搅拌过夜。TLC检测仍有原料(乙醚:石油醚=l:2)。补加丙二酸二乙酯50mg,搅拌反应24h。停止反应。反应液倾入盐酸稀溶液中,乙醚(30mlx3)萃取。有机相用饱和Na2CO,溶液,饱和NaCl溶液洗涤,无水Na2Sq干燥,除去溶剂。制各型板层析分离(石油醚:乙醚=2:1),得产品293mg(MW368.5),产率79.6%。4.2化学计量的V用于charlton的Michad加成:称量V(MW369.5,185mg,O.5mm01),溶于无水THF(10mb中。抽真空,充N,气保护下,加入L认lH‘∞w37.5,19mg,O.5ret001)。冰浴冷却下,搅拌0.5h。称量亚苄基苯乙酮(MW208.3,208mg,lmm01),丙二酸二乙酯(MW160.2,192mg,1.2mm01),加入反应体系中。TLC跟踪检测反应,36h后,仍有少量原料。使用制备型层析分离(石油醚:乙醚=2:1),得产品184mg,产率为50%4.3化学计量的V用于环己烯酮的Michael加成:称量V(MW369.5,18$mg,0.5mm01),溶于无水THF(10mi)中。抽真空,充N2气保护下,加入LiAlI{I(MW37.5,19mg,0.5ret001)。冰浴冷却下,搅拌O.5h。称量2,3.环己烯酮(MW96.1,96mg,Imm01),丙二酸二乙酯(MW160.2,192mg,1.2ret001),加入反应体系中。其余步骤同4.2(催化剂不同,用量不同,反应时间不同)。使用制备型层析分离(石油醚:乙醚_2:1),得产品(MW256.3)16ling,产率为62.8%。25
4.4VII用于催化charlton的Michael加成:称量VH(MW313.4,174rag,0.5mm01),溶于无水THF(10m1)中。抽真空,充N2气保护下,加入LtAlH4(MW37.5,19mg,O.5mm01)。冰浴冷却下,搅拌O.5h。称量亚苄基苯乙酮(MW208.3,208mg,Imm01),丙二酸二乙酯(MW160.2,192mg,1.2ret001),加入反应体系中,其余步骤同4.2。TLC跟踪检测反应,两天后,停止反应。处理得产品(MW368.5)220mg。产率59.7%。4.5VII用于催化环己烯酮的Michael加成:称量VII(MW313.4,174mg,O.5ret001),溶于无水TI-IF(10m1)中。抽真空,充N2气保护下,加入LiAlH4(MW37.5,19mg,O.5mm01)。冰浴冷却下,搅拌O.5h。称量2,3一环己烯酮(MW96.1,96mg,lmm01),丙二酸二乙酯(MW160.2,192rag,1.2ret001),加入反应体系中,其余步骤同4.3。TLC跟踪检测反应,两天后,停止反应.处理得产品(MW368.5)250rag。产率97.6%。5以亮氨酸为原料,合成了一种新化合物:合成路线及反应式如下:>/≮00H蚤>/、c呲/旭可/i,‘冷NHc022Mc⋯卿寺y丫C玺Ph甚c-昔>/飞c镏Ph26盎硼淄
重墼兰至圭竺堡C}bCl(cH3CH2)3NPh>/o嚣Ic=O>\也P伽h、PhX(1)亮氨酸甲酯化:无水甲醇50ml,冰浴冷却至((-50c)。搅拌下,滴加二氯亚砜7.9ml。份加亮氨酸(MWl31.0,13.19)搅拌0.5h,水浴加热,升温至400C,搅拌反应2h,真空薄膜旋转仪旋干。乙醚洗涤反应产物,弃去上层清液。减压蒸馏,600(2,5mmHg得产品11.759(MWl45.o),产率8l%。(2)亮氨酸甲酯与溴苯格氏试剂反应。溴苯格氏试剂的制备同前。称量亮氨酸甲酯(MWl45.0,2.Og),加入反应体系中。室温搅拌0.5h后,加热回流2.5h。TLC跟踪检测反应进程。停止反应。向反应液中,加入冰水。NH,CI溶液调中性。乙酸乙酯(20mlX3)萃取。无水NaSo.干燥,蒸去溶剂。得氨醇产品(熔点134.135"12)3.1549,(MW269),产率85%。氨醇与苯酰氯反应:称量氨酵(MW269,0.29),溶于5ml二氯甲烷。加入三乙胺O.1ral。对应称取苯酰氯(MWl40.5,105nag),加入反应体系。搅拌反应5h(TLC检测反应进程),停止反应,加入稀盐酸。二氯甲烷(20mlX3)萃取水层。合并有机相,无水NaSoI干燥,蒸去溶剂。得产品(MW373,216rag)产率78%。红外鉴定。氨醇与乙二酰氯反应:称量氨醇(MW269,100rag),溶于10ml二氯甲烷。加入三乙胺0.1ml。对应称取乙二酰氯(MW,26mg),加入反应体系。搅拌反应过夜。TLC检测,仍有反应物.补加2eq的乙二酰氯.搅拌反应2h,停止反应,加入稀盐酸2ml。振荡后,加入等体积的水。二氯甲烷(30ml×1)萃取水层。有机相用无水NaSo.干燥,蒸去溶剂。用丙酮溶解,冷冻结晶,得产品(MW592,167mg),氢谱鉴定。6以青藤碱为原料,进行了一系列改性研究。青藤碱,英文名为Sinomenine,分予式为C.9IkN04,来自于防己科植物青藤茎和根,蝙蝠葛的叶。青藤碱是针状结晶,熔点1610C,易溶于乙醇.丙酮.氯仿和稀碱中,在水.乙醚和苯中溶解度较小。青藤碱是生物碱的一种。属于吗啡类药物。具有明显的镇痛作用。镇痛剂量为吗啡的10倍,持续时间较短。并且具有降压.消炎的作用。青藤碱类药物已用于治疗风湿性关节炎和神经病。因此我们以青藤碱为母体,对其进行了改性研究,以其获得药理活性较好的化合物。27淄
:/天津大学硕士生论文青藤碱水解得到二酮化合物,并将二酮化合物与二胺反应。本实验路线设计如下。青藤碱及其二酮的结构式如下:Oo{3nlw出oBTHF+№&—=可■一o,强卅》哪格氏试剂包括溴苯.正溴丁烷.溴乙烷.异溴丙烷.苄氯的格氏试剂。产物包括:Oa龟X¨CH0Q{3XVoCH3ⅪU28O(1置3XⅣoCH3XⅥ
天津大学硕士生论文二酮与乙二胺.邻苯二胺及羟胺等反应,得产物XVII.XVIII.XIVoc蝎+f,m垄醛:苎。嘶+l、m垒卫c》⋯@三一oc》c屿c强c均矿喝c琊~c蝎催::N.c蝎Ho∥√NOHXD(6.1青藤碱与溴苯格氏试剂反应:(1)溴苯格氏试剂,见前文。(2)称量青藤碱(MW329.38,0.59),溶于无水n{F。搅拌,N,保护下,加入溴苯格氏试剂(MW181.4,1.0019)。反应体系颜色由浅黄色变为黄色浑浊液。颜色逐渐加深至透明棕黄色液体。TLC跟踪检测反应进程(乙酸乙酯:甲醇:三乙胺=8:0.4:0.2)。反应过夜。TLC检测,反应完毕。后处理。反应液中加入冰水,氯化铵溶液调至中性。氯仿萃取(3x30m1)。萃取液浓缩后,硅胶吸附。柱分离(乙酸乙酯:甲醇:三乙胺=8:0.2:0.05)。蒸去溶剂,真空干燥,得产品Xll436mg,产率70.6%。为了鉴定其结构,我们培养了单晶,用X—ray确定了其绝对构型。并且测定了其红外,及。HNMR,13CNMR。
天津大学硕士生论文6.2青藤碱与溴乙烷格氏试剂反应:(1)溴乙烷格氏试剂,见前文(2)称量青藤碱(MW329.38,39),溶于无水THF。冰浴冷却至00C。搅拌,N,保护下,加入溴乙烷格氏试剂(MWl33.3,4.8569)反应现象同6.1,TLC检测反应进程。反应32小时后,TLC检测(乙酸乙酯:丙酮:三乙胺=l:l:O.2),反应完毕。后处理同6.1。硅胶吸附后,G-3砂芯漏斗分离(乙醚:三乙胺=lO:1)。TLC检测一个点。浓缩,真空干燥,得产品XIII3.1259。产率为95.5%。红外,‘HNMR,初步鉴定其结构。6_3青藤碱与溴丁烷格氏试剂反应(1)溴丁烷格氏试剂,见前文(2)称量青藤碱(Mw329.38,0.59),溶于无水THF。冰浴冷却至00C。搅拌,N保护下,加入溴丁烷格氏试剂(MWl61.4,0.9799)。其余同反应6.2。(不同的反应条件及不同的试剂,反应时间不同)。反应完毕后,TLC检测一个点。浓缩,真空干燥,得产品XIV0.5889。产率为97.3%。红外,1HNMR,鉴定其结构。6.4青藤碱与异溴丙烷格氏试剂反应(1)异溴丙烷格氏试剂,见前文(2)称量青藤碱(MW329.38,0.59).溶于无水11硬。其余同实验6.2。加入异溴丙烷格氏试剂(MWl47.3,0.2249)。反应完毕后,TLC检测一个点。浓缩,真空干燥,得产品XV0.5679。产率为96.6%。红外,1HNMR,”CNMR鉴定其结构。6.5青藤碱与苄基氯格氏试剂反应(1)苄基氯格氏试剂的制各同其它格氏试剂,只是制备时用无水乙醚代替无水THF(2)称量青藤碱(MW329.38,lg),溶于无水THF。其余同实验6.2。加入苄基氯格氏试剂(MWl51.0,0.4589)。反应完毕后,TLC检测一个点。浓缩,真空干燥,得产品XVl0.8479。产率为66.2%。红外,1I-INMR鉴定其结构。6.6青藤碱的二酮与乙二胺反应称量青藤碱的二酮(MW314.6,0.59),溶于乙醇中。室温搅拌下,加入乙二胺(MW60.1,143mg,1.5eq)。搅拌反应,瓶壁上出现白色沉淀。TLC跟踪检测反应进程(乙酸乙酯:甲醇:三乙胺=8:0.4:0.2)。反应1h后,TLC检测,反应完毕。停止反应,后处理。向反应液里加入苯,静置,过滤,干燥,得产品XVll533mg。产率为98.6%。红外,1HNMR鉴定其结构。6.7青藤碱的二酮与邻苯二胺反应称量青藤碱的二酮(MW314.6,O.59),溶于乙醇中。室温搅拌下,加入邻苯二胺(MW108.2,172,leq)。搅拌反应,瓶壁上出现白色沉淀。TLC跟踪检测反应进程(乙酸乙酯:甲醇:三乙胺=8:0.4:0.2)。反应过夜。TLC检测,反应完毕。停止反应,后处理。向反应液里加入苯,静置,过滤,干燥,得产品XVIll400mg。产率为64.8%。红外,1HNMR,鉴定其结构。6.8青藤碱的二酮与羟胺反应称量青藤碱的二酮(MW314.6,O.59),溶于乙醇中。室温搅拌下,加入羟3O
重童查兰堡圭皇丝兰胺(MW69.5,243,2.2eq)。搅拌反应,瓶壁上出现白色沉淀。TLC跟踪检测反应进程(乙酸乙酯:石油醚:三乙胺=l:1:O.2)。反应4h后,TLC检测,未反应。补加leq羟胺。反应过夜。TLC检测,反应完毕。停止反应,后处理。向反应液里加入苯,静置,过滤,干燥,得产品XIX530mg。产率为96.2%。红外,1HNMR鉴定其结构。各注:我们用X-ray确定了化合物xⅡ绝对构型。对化合物XIII-XIX只通过红外、1HNMR、nCNMR初步确定了其结构。但我们已经纯化了产品XIII-XIX,正在培养单晶,准备进一步测定X—ray、NOE进一步确定其绝对构型。31
天津大学硕士生论文第三章实验讨论及结果分析第一节实验讨论本文对氨基醇的合成及不对称Michael加称反应进行了尝试性研究:青藤碱是~类具有强镇痛类吗啡类药物,本文试图对其进行改性研究以增强其药效。下面将实验路线及实验中出现的问题分别进行讨论。1.脯氨酸与B一萘酚甲酯化反应c02HOHIIIC02CH3oHC=OIo产物l,在二氯亚砜作用下,与过量的甲醇发生酯化反应。本反应可能有两种产物II和III。III为内酯化产物。据.1amesH.short在文献“Theu∞ofAminoAcidintheMannichReaction"’所提及的方法,我们用苯作溶剂,I和二氯亚砜(200mg:0.2m1),用加热套直接加热回流反应。因产物I溶解度不好,导致炭化。第二次仍用苯作溶剂,改用油浴间接加热,但反应结果同上。二次失败后,我们放弃了文献所用方法,而改用甲醇作溶剂,相比较而言。甲醇比苯有如下优点:(1)甲醇沸点较低,可避免反应物炭化(2)甲醇对l溶解性较好:缺点:(1)甲醇作为反应物与l成酯(2)甲醇与二氯亚砜作用,生成硫酸二甲酯。换用甲醇作溶剂后,首先在一5。C下,滴加二氯亚砜,后水浴加热至400C,保温反应2_3h。尝试了几次,但产率都不高。最后用回流代替保温40。C后,取得了意想不到的结果,产率达到94%。产品用红外,1HNMR检测后,确定为11.I首先和二氯亚砜反应生成酰氯。酰氯比较活泼,易生成动力学稳定产物II。且一旦生成II,较难发生可逆反应,从而生成nI。32对蹬∞2∞
天津大学硕士生论文2.合成配体的后处理中温度的影响H与Grinard试剂发生格氏反应。格氏反应要求无水无氧且低温反应。我们在合成配体的过程中,温度始终控制在室温以下(反应开始为(00C,后期为室温)。但反应完毕后,真空旋干溶剂过程中,温度偏高,导致产物分解。后处理前,反应液TLC检测,是一种产品,后处理后:TLC检测发现变为两种产品。尤其是溴丁烷配体合成时,反应液后处理变为两种产品,柱色谱分离,旋干溶剂后。真空干燥,氢谱发现有不明峰存在。在以后的产品处理时,温度都控制在500C以内。产品作氢谱时,仍因为样品加热而导致不明峰出现。直至第三次才得到满意的数据。我们认为不明峰,可能时配体上叔羟基受热不稳定,发生消除反应。3.配体Ⅵ合成后,放入冰箱,冷冻备用。没有经‘HNMR鉴定其结构。原因是反应液后处理后,有十二烷在产品中,真空旋干及减压蒸馏都不适合分离。因此只能采用柱色谱分离,待做。4.配体与LiAIH4络和,催化Michael加成反应机理:拶A旗+oEt5.配体用量对反应有一定影响。化学计量催化剂的效采建化量的效果好。如用溴丁烷配体,0.3eq时,∞%值为lO.46,当用O.5eq时,ee%值则增至35.4。设想如果采用2—3个当量(文献参考剂量),效果会更好。催化4.1与4.2相比,4.2的收率较底。可能的原因:制各型板层析分离,会导致部分产物浪费,无法收集;而且乙二酸二乙酯的当量数,4.1比4.2用的多。6.杂环双金属络合物用于不对称合成,在国内起步较晚,报道较少。本文着重进行了此方面的研究。其中化学计量的溴丁烷配体催化效果最好。当使用3■、●/Oo260XVⅡICuIl26N302388498.6+2365>260XⅨCl8lj【24N3043464962+2942262(257炭化)36
天津大学硕士生论文2.谱图解析2.1化合物I的图谱解析(a)化合物I的1脚伽t谱表1protonsppm积分面积7.168.8.230(d/t)4.850(S11.026(S)4.061(q)3.373(m)2.532-2.151㈤2.150(m)6H2H1H2HCab注在fy=4.650及6=3.300附近为CD30D峰。Ha的峰与—0D峰重合在一起,出现在6叫.851附近。Hd受Hf偶合,裂分为四重峰。原因在于Cd为手性碳原子,正是由于这个不对称碳原子的存在,使其附近的亚甲基上的H化学环境不同,它们是不等价的,因此分别和Hd偶合,Hd裂分为四重峰。萘环上氢H4、H5为三重峰,而Hl、H2、H3和H6为二重峰。(b)化合物I的红外谱图表2基团C=O0—H苯环oH苯环骨架C—N苯环C—H振动类型伸缩振动面外弯曲振动2.2化合物Ⅱ的图谱解析化合物II的1瑚讯m谱表3吸收频率1739.7883219.3433038.0611619.6021335.486756.094,707.094积分面积H广H6HaHeHdHe,HeHf’Hf,HgHb7.126-7.856(d/t)4.038:(q)3.769(s)3.476(q)3.132,2.424(q/m)2.299"2.050(m)1.884“n)1.287(br.)376H2H3HlH2HlH^kHg挑№~m一~№h。吣m。m。
墨兰查兰堡圭圭兰兰Ha裂分为四重峰,的氢萘环上氢H.、H,为和H。为二重峰。分为四重峰。原因在于Hf以Hd裂分为四重峰。同Ha一样,Jl=J2=6.4。2.3化合物VII的图谱解析化合物VII的1HNMR谱表4Jl=14.4,J2-14.0。萘环上为三重峰,而Hl、H2、H3Hd和Hf.Hf、偶合,裂的两个氢是不等同氢,所积分面积Hl—H6HaHbHe,HeHf,HcHg,HhHi7.196-7.740(d/t)4.231(q)2.673m2.789,2.144(q/m)1.648-1.178(m)1.648.I.178(m)0.797(m)6H2H1H2H3H6HHb同样受到Cf上两个化学不等价H质子的偶合,裂分为四重峰。Ha裂分为四重峰,如右图所示Jl=J2=14.8。核磁共振显示,含有杂质三乙胺。在5=2.380和8=0.948分别为三乙胺上亚甲基和甲基的吸收峰,满足H的个数比。8=7.162为溶剂(C6D。)峰。经计算He,He’的偶合常数J。=6.4,J2=6.4,具体见附图。2.4化合物v的图谱解析化合物V的1I-1NIVIR谱表S积分面积H广H6HaHbHe,He’Hf,Hg,Hh7.256-7.839(d/t)4.336(q)2.806(02.895.2.230(m)1.648—1.178(m)6H2HlH2H22H谱图上在8=7.229处为c。D6的吸收峰。谱图在8=2.380处C出现了杂质三乙胺的亚甲基吸收峰,而甲基则和Hg、Hr.Hf的的吸收峰混合在一起。在此图中He,He’裂分为六重峰,分别先后与Hg及自身氢发生邻碳和同碳偶合。经计算偶合常数J。=10.8,J2=10。见附图。38
天津大学硕士生论文青藤碱用硼氢化钠还原产物的图谱解析(a)还原产物的1HNMR谱表6protonsppm积分面积H12H.HlH。(O-H)H"H6H13坞H16HloH”H14HIs(b)还原产物的红外谱图表7O(】】13lS19C随地与H6之间的邻碳偶合,据Karplus方程式,联系谱图。得出由于空间构型的影响,心只与比上的一个氢偶合,裂分为二重峰。巳为手性碳原予,受其影响,其邻近的c,上的两个氢为不等价核。其中一个与H6邻位偶合,同时也上两个氢核相互偶合,因此地上两个氢核裂分为二重和四重峰。而且地上两个氢核还受到远程偶合,每个峰又裂分为两个小峰。计算偶合常数,地的偶合常数为Jl=14.8。一恤㈣(如H。的偶合常数(如凡口口丑右图所示。Jl=14.8;Ji=J2=12.8。根据Karplus方程式,Hl和H14二面角为90。C,相互不偶合。因此H。为单峰。同时}19和H。.空间构型上处于相互垂直的位置,因此H¨为单峰,且和HJ,的峰重合在一起,出现在5=1.984。Hl,H2相互偶合,39撕mmm姒m姒m俎俎疆mⅢ㈣一一㈣一一一蜊~删删㈣喇一一一一一一一一~一一一一
耋兰奎耋塑圭兰坚裂分为四重蜂,偶合常数为J。=8.4,J2=8.0。H。,和H。。都发生同碳偶合,裂分为四重峰。H。。上的一个氢还发生了远程偶合,裂分为两个小峰,需通过双照射去偶,进一步确定。还原产物经X-ray测定了其绝对构型,如上图。2.5化合物XlI的图谱解析(a)化合物XII的1HNMR谱表8protons积分面积A卜HHL2H.(O—H)H|HJ,HIsH197.239-7.369(m)6.713(q)6.325(br.)4.712(s)3.846(s)3.381(s)2.071(s)5H2HlHIH3H㈣化合物XII的红外谱图表9(c)化合物XII的13CNMR谱表lO积分面积156.016147.22l145.09l144.440130.08l127.897126.395125.032124.016116.8924019C嚆OCH3lR坨坨坨屺坨妃屺扼圮坨qq国%a&例∞%q
天津大学硕士生论文C,108.8921CC。97.622ICC17,C1874.491,76.357ICC,。57.9661CC。55.9361CC.。54.5241CC.,50.145ICC.。47.9281CC。44.3761CC.。42.1961CC.。35.737lCC。24.4651C化合物XII的1I-INMR中,H。,:邻碳偶合,裂分为四重峰。比的羟基吸收峰为一宽峰。ppm=6.325。化合物Xll的”c"hq~jIR中,q,c。2,Ca因其化学位移值比较接近,考虑到计算误差的存在,所以我们认为其Interchangeable。同样ppm在124.06—130.081之间的C核同上。2.6化合物XIII的图谱解析(a)化合物XIII的1I-INMR谱表11积分面积6.598(034.339(s)3.772(s)3.412(s)2.469(s)1.650(m)2HlH3H2H19C珏化合物XIII的1RNMR中,Hl”邻碳偶合,裂分为四重峰。ppm=7,250处为氘代氯仿的溶剂峰.H∞,H2l的吸收峰出现在ppm=1.650,ppm=0.790处。而且H:.受H。的偶合,裂分为三重峰。偶合常数Jl=7.296,J2-7.374。(b)化合物XIII的红外谱图表124l
茎兰查耋堡圭圭兰兰2.7化合物XIV的图谱解析(a)化合物XIV的1HNMR谱表13protons积分面积Hlj6.693(q)2HH(o-H)6.213(s)1HHB4.310(s)1HH173.814(s)3HHI。3.429(s)3HH192.726(s)3H些Q望:!!生!:!!!f型塑oaH31819C}b化合物XIV的1ItNMR中,H1,2邻碳偶合,裂分为四重峰。ppm=7.249处的吸收为氘代氯仿的溶剂峰。㈣化合物XIV的红外谱图表142.8化合物XV的图谱解析∞化合物XV的1I:INMR谱表15protons积分面积Hlj6.621(q)2HH84A02(s)1HH”3.783(s)3HHll3.401(s)3H,。H192.497(s)3He屿H201.225(s)IHc22rbH,.O.948(d)3H生旦:!垫盟!旦4219C珏
耋兰奎兰矍圭兰!兰(b)化合物XV的红外谱图表16(c)化合物XIV的”CISlMR谱表17carbons积分面积157.900144.918144.156130.687125.237118.818108.63496.55974.839-76.3157.67755.88754.17647.78044.11942.34838.83l36.51734.77824.34417.60615.55619C砖化合物XV的1HNMR中,H。,:邻碳偶合,裂分为四重峰,不太明显。ppm=7.238附近为溶剂氘代氯仿的吸收峰。异丙基上的R,,H。受H。的偶合,裂分为二重峰,偶合常数分别为6.802和6.736。化舍物XV的”CNMR中,C,,C。:因其化学位移值比较接近,考虑到计算误差的存在,所以我们认为其Interchangeable。C16,C9,C6,以及ppm在47.780—34.778之间的C核同上。43圮坨圮坨屺坨圮圮坨坨坨屺圮坨坨坨坨忙忙坨圮‘6DD踟D踟DD&踟DD&踟∞踟踟‘j0j踟白GD
天津人学坝l生论义2.9化合物XVI的图谱解析(a)化合物XVI的1HNMR谱表18Interpretppm积分面积H2l7.199(05HH”6.631(m)2HH。6.325(br.)1HH84.402(s)1HH,,3.782(s)3HH】83.496(s)3HH。92.498(s)3H(b)化合物XVI的红外谱图表19oo-13182.10化合物XVII的图谱解析(a)化合物XVII的1HNMR谱表20Interpret积分面积H202I7.591(t)2HH¨6.598(s)2HH46.165(s)1HH.,3.692(s)3HHf92.705(s)3H图潜巾,5=7.242处为CDCI,溶剂峰。19C卜b
天津大学硕士生论文(b)化合物XVII的红外谱图表2l2.11化合物XVIII的图谱解析(a)化合物XVIII的1HNMR谱表22积分面积H2I7.854((t)2HH:o7.599(02HHlj6.579(s)2HH|6.21Ks)1HH173.679(s)3HH192.599(s)3H(b)化合物姗的红外谱图表23化合物XVIII的1HNMR谱中,H。,2为单峰。H的羟基吸收峰在ppm=6.211。H:o,H2I相互偶合,化学位移值分别在7.854,7.599。H:。裂分为三重峰。ppm=7.239处为溶剂峰。45
2.12化合物XIX的图谱解析(a)化合物X1X的1HNMR谱表24积分面积H202l8.246(s)2HHl26.627(s)2HH。6.505(br.)1HH”3.748(s)3HH192.687(s)3H琶鞋"-N一‰“。冬出。。s一‰如一删薪_6图谱中,8=7.245处为CDCI,溶剂峰。H.的吸收峰N02H0为一宽峰。(b)化合物XIX的红外谱图表25·2.13化合物Ⅺ的图谱解析化合物XI的红外谱图表26笨环C—H的而外弯曲振动的两个吸收峰是苯环单取代的特征吸收。
塑丝矍圭兰丝三——2.14化合物IX的红外谱图表27基团振动类型吸收频率强度N—H苯环C—HC--OC=CN-】HC—N苯环C-H伸缩振动弯曲振动倍频伸缩振动弯曲振动伸缩振动面外弯曲振动3406.2923061.3293031.6071632.4531578.9621527.0151161.340745.417,704.059苯环C—H的面外弯曲振动的两个吸收峰是苯环单取代的特征吸收。2.15青藤碱硼氢化钠还原产物的单晶X-ray衍射数据耋垫生翌!塑!璺璺垒!竺壁!塑!!塑!!堂里!型!坐塑匣!IdentificationcodeEmpiricalformulaFormulaweightTemperatureWavelengthCrystalsystemSpacegroupUnitcelIdimensionsVolumeZDensity(calculated)AbsorptioncoefficientF(ooo)CrystalsizeThetarangefordatacollectionIndexrangesReflectionscollectedIndependentreflectionsCompletenesstotheta225.61AbsorptioncorrectionMax.andmin.Wansmissionp21C19H25N04331.40293(2)K71.073pmOrthorhombicz2(i)2(1)2(1)a=773.83(15)pmb=1013.1(2)pmc=2153.5(4)pm1.6883(6)nm341.304Mg/m30.09Imm。l712ct=907B=907T=9070.4x0.4x0.3mm31.89t025.6l一9<--h<--9,一12<-k《=0,-26<=1<=2655833093【R(in0;00660]97.6%None1.133and0.93l47smwomsms
Re‰ementm锄odFuu.ma慨l删咖uarcs0nF2Data/restraints/parametersGoodness.of-fitonF2FinalRindices【l>2sigma(1)】Rindices(alldata)Absoluteslru血lreparameterExtinctioncoefficientLargestdiff.peakandhole3093/0/218I.018RI=O.0557.wR2=0.1489RI=0.0604.wR2=0.15271.3(14)0.08400)0.217and-0.257c.-3表29Atomic∞ordinat髂(I104)andthermalparameters(pm2x10-1)xYzu(eq)c(1)4481(3)4035(2)11253(1)54(1)o(1)6885(2)2680(2)12575(1)74(1)Cf2)4749(3)3371(2)11804(1)54(1)∞)9412(2)3867(2)11965(1)72(1)C(3)6399(3)3324(2)12044(1)54(1)“319139(2)6834(2)12220(1)66(1)cf4)7764(3)3952(2)11730(1)52(1)鲋)7903(3)9062(2)11615(1)71(1)C(5)i0350(3)6039(2)11219(1)51(1)C(∞9711(3)7205(2)1t605(1)55(1)“718318(3)7961(2)11278(1)54(1)C(8)7673(3)7617(2)10726(1)53(1)C(9)6915(3)5809(2)9953(I)53(1)C㈤5352(3)5391(2)i0340(1)56(1)C(II)57830)4690(2)10943(I)48(1)C(12)74750)4688(2)1I190(I)47(1)c(13)8961(3)5339(2)10828(I)47(1)cfl4)8270(3)64.13(2)10384(I)49(1)c㈣97970)4259(2)10419(i)55【1)c(1618537(4)3730(2)9935(1)6l(1)c(17)5612(4)2007(4)12913(I)87(1)州17)7767(3)4803(2)9571(1)59(1)c㈤6737(5)9974(3)11332(2)95(1)业2型盟一竺盟——型塑L——旦L一48
耋塞查兰堡圭竺篓兰表30Selectedbondlengths(pm)and):BondDjsLo(I)一c(3)o(1)一c(17)o(2)一c(4)o(3)一c(6)o(4)一C(7)o【4)一C(18)N17)一c(9)N(17)一c(16)N(17)一c(19)c(6)一c【7)C(7)一c(8)q8)一c(14)c(10)一c【11)c(12)一c(13)136.9(3)14n2(3)137.5(3)144.5(3)136.8(3)142.7(4)146.7(3)146.6(3)145.9(3)149.8(3)133.“3)149.8(3)151.7(3)153.7(3)O(1)一c(3)一c(2)o(I)一C13)一C(4)C(3)一O(1)一C07)o(2)一C(4)一c(3)o(2)一C(4)一C02)o(3)一C(6)一c(5)O(3)一c(6)一c(7)o(4)一C(7)一c(6)o(4)一C(7)一C(8)C(7)一o(4)一q18)N(17)一C(9)一COO)N(17)一C(9)一C04)c(9)一N(17卜c(16)c(12)一c(13)一q15)125.8(2)114.3(2)118.2(2)1194(2)119.2(2)113.4(2)110.o(2)109.7(2)126.6(2)l16.8(2)118.o(2)108.1(2)I13.5(2)107.4(2)49
重量奎兰堡圭兰篁兰;;—一2.16青藤碱与溴苯Grignard试剂反应产物的单晶x—ray衍射数据表3lAtomiccoordinates(x104)andthermalparameters(pm2x10一1)U(eq)isdefinedasonethirdofthetraceoftheorthogonalizeduijtensor5O善:丽㈣㈣㈣㈣㈣㈣㈣删㈣㈣姗㈣㈣㈣㈣㈣㈣㈣㈣㈣㈣㈣嘞㈣㈣㈣㈣㈣㈣一:一一一一一一一一一一一一一一一一一一一一一一一一一一一一一一一,一㈣一一一一一一㈣一剐一一一一一一一一一一一一圳一一一一一一。一一㈣一一一一一一一一一一一㈣㈣嗡一㈣嘲一一一一一一一一一一一j...;一、,、,、,、,、,、,、,、,、,、,、,、,、,、,、,、,、,丌动"q”动”q∞旬D酌∞m¨坦BH“M""博坶加孔挖幻M筋可叫叫叫qqq嘶qqqqqqqqqqqqMqqqqqqqqq
Fig.IViewofIIdrawnwith30%probabilityellipsoidsBondDist.Angle。O(1)一C(3)138.6(4)o(I)一c(3)一c(2)1257(3)O(I)一c(19)139.5(4)o(1)一c(3)一c(4)113.9(3)0(2)一C(4)1358(4)C(3)一o(1)一c(19)1171(3)O‘3卜C(6)144.7(3)0(2)一C(4)一c‘3)1200(31O(4)一C(7)137.8(3)o(2)一C(4)一C(12)1204(3)O(4)一C(18)1410(3)O(3)一C(6)一c(5)1ill(2)N(17)一C(9)1488(3)o(3)一C(6)一C(7)1087(2)N(17)一C(16)1477(3)O(4)一C(7)一C(6)109.3(2)N(17)一C(17)1474(3)O(4)一C(7)一C(8)1260(3)C(6)一C(7)1509(4)C(7)一O(4)一C(18)118I(2)C(7)一C(8)131.9(4)N(17)一C(9)一C(10)118.0(2)C【8)一C(14)150.414)N(17)~C(9)一CIl4)108I(2)C(10)一C(1I)I522(4)C(9)一N(17)-C(16)1145(2)C(12)一C《13)1547(4)C(12)一C(13)一C(15)1082(2l5
重量奎兰堡圭圭兰圣第四章结论参阅有关文献,本文对手性氨基醇的合成及不对称催化的Michael加成反应进行了研究与探讨。并以青藤碱及其二酮为原料,进行了改性研究,合成了一系列具有药理活性的衍生物。得出结论如下:1.以脯氨酸、B一萘酚为原料合成了I,产率为90.4%;合成了化合物Ⅱ,产率为82%:合成了IV-VIII,收率依次为94.2%、92.7%、85.1%、90.O%。均为新化合物,进行了结构鉴定。2.首次报道了手性氨醇v和VII用于催化丙二酸二乙酯与环己烯酮或亚苯基苯乙酮的不对称Michael加成反应的结果。3.以青藤碱及其二酮为原料,合成了化合物XII-XVlII,均为新化合物。其结构经元素分析、红外光谱及核磁共振谱的鉴定。我们还通过X-ray确定了化合物XII以及青藤碱的硼氢化钠还原产物的绝对构型。
重冀查兰堡圭竺篁兰参考文献【l】ErasmusM.v0四;HaraldCJrGger;andMasakatus.Shibasaki:Angew.Chem.Int.Ed、1999,38,1570-1577『21Koiehi.Yamakoshi;SimonJ.Harwood;Motoum.Kanai:TettahedronLetters40,‘1999,2565-2568【3】MasakatusShibasaki,Hiroaki.Sasai;TakayoshiAmi:Pure&Appl,vol70,No.5PP1027-1034,1998【4】G.Manickam;G.Sundararajan:Tetrahedron,55,1999,2721-2736【5】ShingoYamaski;Takehikolida;Masakatsushibasaki:Tetrahedron,55,1999,8859-8867『61HiroakiSasai;TakeyukiSuzuki;NorileItoh;Masakatsushibasaki:TetrahedronLetters,、,b1.34,pp851-854,1993f71G.Manickam&G.Sundaramjan:hdianJoumalOfChemistry:、b1.36,June,J99Zpp516_-518【8】HiroakiSasai;TakeyoshiA_rai;YoshinoriSatow;K.N.Houk;MasakatsuShibasaki:J.Am.Chem.Soe.1995,ll7’6194__6198[9】MasahikoiYamaguchi;TaiShiraishi;MasahiroHirama:J.Org.Chem.1996,61,3520一3530[10】TakehikoIida;NoriyoshiYamamoto;ShigekiMatsunaga;Hee-GweonWoo;MasakatusShibasaki:Angew.Chem.Int.Ed.1998;37.NO.16【ll】G.Manickam;G.Sundararajan:TeU"ahedron,Asymmery,vol8,No.13,pp227l—2278.1997【12】ErikKeller,NoraVeldman;AnthonyL.Slack;BenL.Feringa:Tetrahedron,Asymmery,Vol8,No.20,pp3403—3413B,1997『131BerechnetnachEw.Kfister:Helv,33,590(1950)【14】JamesH.Shoa(1);C.WayneOurs:J.OfHeterocyclicChem.;Oct.1975【15】EDGARw.GARBISCH;JR.:JOURALOFOrganicChemistry,Volume30,namber7,,钾7f161DRARTHURKOTZ;GoTTINGEN;PatentiertimDeutschemReiche,Vom16,November1907曲『17]M.S.Kharaseh;G.Sosnovsky:JOC,1322-1326,,卵乎【18】樊能廷,有机合成事典:北京理工出版社1992,l【19】YukioHitotsuYanagi;HiroshiIkuta;KuninikoNishimura;KoichiTakeya;HidcjiItokawa:J.Chem.Soe.Chem.Conl//lun.,1994,lI’2,,,访[20】IkuoIiiima;J衄一ichiMinamikawa;ArthurE.Jacobson;ArnoldBrossi;KennerC.Ricc:J.Org.Chem.,V0143,NO.7,1978【21】YukioHitotsuYanagi;KanihikoNishimura;HkoshiIkuta;KoichiTokeya:J.O璀.Chem.,1995,60,4549-4558『221Ikuolijima;Jun-ichiMinamikawa;AYthurE.Jacobson;JournalOfMedicalChemistry,1978,vol2l,No4[231D.S.WULFMAN;C.F.C00PER;SynthesisCommunications,924,圩77『241MiehaelG.B.Drew;LaurenceM.Harwood;GyoosoonPark;DavidW.Price;
天津大学硕士生论文Simon.N.G.Tyler;ChanRyangPark:;SooGyeongCho:Tetrahedron57(2001)5641-5648【25】TamioNishimura;KojiKitajima:J.Or晷Chem.,Vol,44,No.5,1979【26】AchimHene;StevenV.Ley;KieronE.Wesson:J.Chem.Soc.,PerkinTrans.1,1997【27】OkabeK.ReductionProductofSinomenine.Yakugak-uZasshi,1962,82:1496~1502【28】柯铭清:中草药有效成分理化和药理特性194,1980【29】njimaI,MinamikawaJI,JacobsonAEeta1.Studiesinthe㈩.MorphinSeries.J=Org.Chem.,1978,43(7):1462~1463【30]黄宪,陈振初;有机合成化学,化学工业出版社,1981.10【3l】RM.西尔弗斯坦,G.C.巴斯勒;有机化合物的光谱鉴定,科学出版社【32】张正行;有机光谱分析,中国药科大学出版社,1987.9【33】陈茹玉;红外光谱使用指南,1992.4【34】李述文,范如霖;实用有机化学手册,1979.10【35】余仲键,李松兰,张殿坤;现代有机分析,1994.7【36】刑其毅,许瑞秋,周政:基础有机化学,上下册,第二版,北京:高等教育出版社,1994[37】恽愧宏,高鸿宾,任贵忠:高等有机化学,北京:高等教育出版社,1988【38】范如霖;有机合成特殊技术,上海:上海交通大学出版社,旧7岁
附录1化合物I的1Ⅱ舳谱。o。N\cI一\∞口oE:口:1N工oo
附录2化合物I的红外诺图aouel41Ⅲs帅刈暑o£:∞or9I_oooN.罱ja口暑o.^epJnl母∞k∞暑,cm,∞I卜山∞.斗1I一∞一Eo二Q装薅裂。、r棼母ooo∞∞J.也凸《叱.o—m¨椎g幸廿繇销幡扑晕避扑割扑K繁K
一=====善亡1\彳:毫.i·jj,,’7、。、.·。·t-ql".:}:!忙n口.oJd.H‘Tn,忙,∞寸警_u2口z_a暮iqh∞
⋯⋯“⋯⋯⋯⋯~⋯●·●⋯mo“⋯Ⅲ-····⋯⋯tzo⋯q、m⋯d⋯⋯‘⋯-uoLJ一●⋯_∞⋯,Hp⋯£j~⋯●⋯口⋯‘一F一⋯L毫2==’。一=二=拿i0二;暑i附录4化合物VII的1HNMR诺善矗§{=i主=二罟;嚣:;l=i器!i博培E口△1●}●Jfl●●●●r●●●●,LJ。。≈蠢餐o-n¨n-c-:.【3强。:。=§。。£qI强嘏o-oI25矗~=o-西‘§=%}tJl¨HDu》Eu蛊o=_alt口tt卜∞
附录5.:。}。}.’、一Z::j22nl=‘嚣。=呈。’甘盆22一。基蠢lIi‘;-2&。;是;i{弱lIi;&§i{;化合物V的JHNMR谱}———————弓。————≥l。:。dl___‘[,1==主<———========篁主———:====;;jEJ.=====三——1=_-_-___--_;≤)!}:,,=.∞“n●.¨“一H=.“oN.=o_.口∞o.“:._i}1r}.【N竹-£.∞n.__。莨镪圪卜∞亨£∞罟三宝舌三钫
附录62=一£嚣;{巍{{i品嚣S二吧7。’)蓦q=l§§i嚣5;:奄《i萎§萄抟]!}连H}!}!.)。:密
附录7化合物V的1HNMR谱(有杂质)⋯uu5.:0£:一61)i蟮}!々}::Eo口N竹h-._nh.“。圬墙圪■“p口o-o-_pc鬯●nh-o_●●●.f_I-io.●,l●■,tnL王p^x-,H.-L;H.aoo.n口‘-^nn.£--.。-∞-,川--uoo‘。._h‘c.I;.,“.*.。-■。吧ojP_zcpo毒暑ug芒N∞o.o:t三“o●H-●●toHPHnH,口u‘.ul口--.-H__●-●‘u.-J●_Ec善圭;.6.11,^lun=0.矿。酶一:j一护《2转路、矬}{_zcI。o-u_c.三o._∞皇。ottL-to。oN_N,oz。pot;●.2a=兰矗u)Eu∞∞ozd口_t口z《P日j乏
63。nn,了h一—一扣.一心小.~T—r口—一卜—一仃}3札I。.~≮~—一口.n。.。;.。廿丁。—一.:.、卜.卜,krk忙PFrB;Fk[窿P~K窿
附录9青藤碱用硼氢化钠还甄产物的红外诺圈03u口11ImguRLJ■64∞Inn01.100N.No^13f-.^∞刁co乏蘧翳硪~<轱鞋ooon∞卜止口《咩oIB¨臻警
Hdd附录10化台物xII的1皿帅m谱looN\o一\一∞二一
1矾1晒r—一1矾—rr广—一附录ll化合物X11的13cNMR谱£L正;篓黪雅!nJo口臼一
附录13eauelllmsu口刈嗤卜∞K冀o1.NooN.o一^J∞丁c叮,im口23c.L§dso.no皇u=}h∞o弓co}篁nooon∞.Lko叠.o|g。<妻聿世。稚基套导繇林帐扑晕避扑剐扑K辛}K
譬驴f、1i百=Fr_一可强百r了—一1五百rr—一百醑百r了—一Hdd附录14化合物)【|叮的IHNMR谱NooN\l/NN#一
附录15化台物xⅢ的红外谱图a3uellI_su口jj^∞(I,卜Uo矗o∞醉囊基世oo.6时|01.NooN.oI.^J可丁c∞1i∞DsJ3cJ-一ods口.80里譬盎置km{∞童穹乎锑球帐扑摹迟扑剐扑K赞米
附录16化合物XⅣ的l瑚Ⅱm主搿;PPHZ:Z地§2§:£22ZZ生。2Q2§Zi。ll蛆23:圭Z1日§2:Z2£i2~#~~/1\N。。N
a3U口:l:l佃su毫l上t薛囊警世N口n蛇oI,NooN.oI.己∞jc母r.i∞≈sJ3LJ卜(9一d譬声.廿2舌呻c譬嚣口§co>篁.L“∞.∞hh_∞一暑。工u∞ooon∞J-止。矗.018o哥辞诛峨貅尊遮扑酎扑K转帐
的IHNMR谱PP阿≥≥:j—f’z:2322i£;§Zn2i±,蛐2§Zl。ZIa2蔓=<=蔓3.五。LO畦O豆92Z。a口纽2=二黜一z】u1/一。/200一百~了
Nooo广uo\二\Noo一一oo可勺z脚o∞oNo
附录20化台物xV的缸外谱圈a0哪!t懈ut』J■74o‘I,卜。矗o∞醵主g$n苫冀ol_NooN.oI.^JBnuB,蔷p宕JnLLL夺)d盖.己。皇盆暮,■m童co,日l卜山∞.们^_I_∞一告D二。审d|球泳礞扑甚避J士甜巾<£、/
附录21化台物x、,l的IHNMR谱N¨o,1l/Noo—
附录22化台物XVI的红外谱圈藁囊晕芈。冀on.olINooN.ol_^J椰3c町f’.^∞可∞JncJ_一n一导皿.一。墨盆c譬hm丑E,co>叠1L厶∞.∞hhp∞一EoLIuooon∞j.止D《芷.01日o}魏林帐扑睾避扑尉扑K瓣怅
T歼百rT—一耍匿西玉二jlL㈠‘£Tn百r百—一百万可广丁—一百丽百F百—一Hdd附录23£艮乱筹NooN\N\一_I
附录24化合物xvII的红外谱图No西o#l_NooN.o1.^J弼丁c∞『’妄mDSJjuJ—o—a∞口.岫o∞工u∞cq-koq哪己∞》叠_L山盘.∞h-z¨∞一EoIIo吨ooon∞.1Lo歪.018一<婪世。稚基穹乎甜林帐扑掌攫扑唧扑K转帐
附录2s化合物XVlll的IHNMR谱一NN/11/~oo—
附录26化合物XVIII的红外谱圈a姗11IⅢsuom卜■∞N6rNooN.o1.^Jmjcmf’.^仍D价JncJ-一“一B”口.o∞tIu∞c日-■m口Ejcm,日■卜山∞.∞hh.1∞一go工o~<妻幸世ooon∞卜也D叠.01日~稚翠自cb搿袜帐扑摹毽扑尉扑K毒}帐
T丽百r下——一T疆百广丁—一百歼下F丁—一¨dd81z艮t譬她"No{)N\N\一一
aouen!船u¨l生∞n寻o.1I_“oo“.or^J∞3c∞f’im口∞Jn£卜一∞『)4∞口.∞o日‘u∞c日。h∞oE,cm}日}卜t|∞.∞*hu们一E∞£o一、r丧}世ooon∞卜uQ歪.01日¨椎g々壬g妹峨扑甚遽扑割扑K毂联
83o∞一卜N—o—oooN.∞一h∞DEm一盘∞∽.x∞可co=ko县暑c∞毒■*罄州一<彝}犟ooo器上dg∞IrI《奋2∞占H∞一嘴基0廿嚣妹倏扑摹蠼特倒扑K毒}帐
a3u口1{!惦uPq■召∞蛊竹一.9葛oroooN.时NJ9DEmld∞∞i毋口sJ了cJ.‰q山暑o,篁皇∞i三∞{。吾0子覃宣诫嚼扑荦迩扑尉扑K赫帐
附录3l青奠碱一氢化钠还甄产物分子结构圈
附录32化合物Xll的分子结构图
o=‘o
致谢在本论文的完成过程中,我的导师李春葆副教授倾注了大量的心血。从文献的查阅,实验方案的确定,研究思路的拓展到论文的撰写,无一不是在导师的悉心指导下完成的。导师严谨的治学态度、渊博的知识面、忘我的工作精神令我终生难忘,并将使我终生受益。在此我衷心地向我的导师表达我深深的感谢和崇高的敬意。赵春常、郑鹏武、张航、陈谊等在实验过程中给予了很大的帮助,借此机会向他们及在实验过程中给予我帮助的老师、同学一一表示感谢。在此,还要感谢我的爱人,她的理解和支持,使我能够顺利的完成论文。我的家人多年来对我的学习和工作倾注了大量的心血。正是他们无微不至的关怀激励着我,我将永远感谢他们。王超2002年1月'
您可能关注的文档
- dh1718型直流稳压电源故障机理及结构改造
- 国产四缸四排汽300mw 汽轮机本体结构改造设计
- 宁陵县2015年公路路网结构改造(农村公路危桥改造、农村公
- 旧钢结构改造施工方案设计
- 2500m3高炉煤气放散阀的结构改造
- 建设工程结构改造实施流程方案_设计
- 混凝土静力切割施工工艺[结构改造]-
- 【硕士论文】减肥药利莫那班的结构改造.pdf
- 对某综合办公楼结构改造方案探究
- 茉莉素的化学结构改造与其抗肿瘤作用的研究
- 不对称michael加成催化反应的研究青藤碱的结构改造
- 环维黄杨星d的结构改造和生物活性的研究
- GBT51087-2015 船厂既有水工构筑物结构改造和加固设计规范
- 多层混凝土结构改造中钢管扣件支撑架应用
- 钢管混凝土拱桥动态悬吊式主梁结构改造技术
- 海洋天然环肽leucamide+a关键片段的结构改造
- 存款计息及账户结构改造方案说明(2011-5-6)终结版
- 翻车机在适应c80火车机械结构改造的前期分析和后续改造