


- 1.36 MB
- 10页

- 1、本文档共5页,可阅读全部内容。
- 2、本文档内容版权归属内容提供方,所产生的收益全部归内容提供方所有。如果您对本文有版权争议,可选择认领,认领后既往收益都归您。
- 3、本文档由用户上传,本站不保证质量和数量令人满意,可能有诸多瑕疵,付费之前,请仔细先通过免费阅读内容等途径辨别内容交易风险。如存在严重挂羊头卖狗肉之情形,可联系本站下载客服投诉处理。
- 文档侵权举报电话:19940600175。
DNA重组生物实验报告课程名称:_生命科学导论实验课_指导老师:______成绩:__________________实验名称:__基因工程实验__实验类型:__生物实验_______同组学生姓名:__一、实验目的和要求基因工程实验是生物实验中极具代表性的一部分,在此次实验中有如下目的和要求:1、通过本实验了解和掌握含GFP基因质粒的提取和琼脂糖DNA电泳的等十个小实验组合而成的基因工程实验的原理和技术。2、了解生物实验的完整流程,对实验室基本操作规范与仪器设备使用方法得到初步了解,从而建立系统、有条理的实验思路。3、培养实验动手能力,在实践中加强对专业知识的认知与体会,融汇贯通,寓学于用,感受生物学科的魅力。二、实验内容和原理基因工程又称DNA重组技术,是将不同来源的基因按照预先设计的蓝图,在体外构建成杂种DNA分子(又称重组DNA分子),然后导入活细胞,以改变生物原有的遗传特性,获得新品种和生产新产品等。基因工程技术为生命科学的研究提供了有力的手段,同时为以基因工程技术为核心的现代生物技术产业的建立奠定了扎实的基础。基因工程技术建立的标志性实验是:①1972年,美国斯坦福大学的伯格(P.Berg)等人将猿猴病毒SV40的DNA和大肠杆菌λ噬菌体DNA分别进行EcoRI酶切,然后用T4DNA连接酶将两个酶切片段进行连接,率先完成了人类历史上第一个DNA分子的体外重组实验,因此荣获了1980年的诺贝尔化学奖。②1973年,美国斯坦福大学的科恩(S.Cohen)等人在体外构建了含四环素和链霉素两个抗性基因的重组质粒,并将其导入大肠杆菌中,获得了双抗性的大肠杆菌转化子,成功完成了第一个基因克隆实验。上述两大科研成果标志着基因工程技术的诞生。一个典型的基因工程技术包含以下几个步骤:1.目的基因和载体DNA的获取。2.目的基因和载体DNA的限制性内切酶的酶切消化。3.酶切后的目的基因和载体DNA的体外重组连接,以获得重组DNA分子。4.通过细菌转化等技术,将重组DNA分子导入细菌等活细胞。5.转化细胞的筛选鉴定以及目的基因表达的检测。其中目的基因、载体、工具酶及宿主细胞是缺一不可的,因此被称为基因工程的四大要素。实验一含GFP基因质粒的提取目前获取目的基因DNA片段常用方法是:从GeneBank中查得目的基因的DNA序列,根据序列设计引物,通过PCR的方法扩增目的基因的DNA片段。根据目的基因的来源不同,用于PCR扩增的模板DNA,大致有两个来源:①由于真核细胞DNA的基因编排方式,是由内含子和外显子等组成,因此,只能通过提取目的基因的mRNA,逆转录合成出cDNA,作为PCR的模板。②原核生物的DNA其基因是连续的。因此提取原核细菌的DNA,就可用于PCR的扩增。本次实验的目的基因就是来自一种细菌编码的脂肪酶基因。细菌基因组DNA的提取先是用溶菌酶及蛋白酶K裂解细胞,释放DNA、RNA及蛋白质等。然后用苯酚/氯仿处理,使蛋白质变性,经离心去除蛋白质及细菌碎片,DNA和RNA存在于上清水相中,然后用RNase10
分解RNA,酒精沉淀DNA,即可获取目标基因组DNA。实验二琼脂糖DNA的电泳DNA分子是两性电解质,在溶液中形成兼性离子而携带电荷,带电分子在电场中将产生移动,这种现象称为电泳。带电分子在电场中移动的快慢,除受到场强、缓冲液类型及缓冲液离子强度等外界因素的影响外,还受到分子的大小、带电量及分子的形态等自身因素的影响。为避免断电后电泳分开不同DNA分子的迅速扩散混合,DNA电泳常采用琼脂糖的凝胶电泳。实验三PCR扩增目的基因1985年美国Cetus公司的穆利斯等人设计并研究成功了一种体外核酸扩增技术PCR(PolymeraseChainReaction),从而荣获了1993年诺贝尔化学奖。这种聚合酶链式反应是一种类似于细胞内DNA的天然复制过程,利用半保留复制的原理,以待扩增的DNA为模板,在体外由引物介导的酶促合成特异的DNA片段。将目的基因DNA在高温(94℃)下解链成为单链模板;人工合成的一对与目的基因两侧序列相互补的寡核苷酸引物在低温(40~60℃)下分别与变性的目的基因片段两侧的两条链的部分序列互补结合;在中等温度(65~75℃)下由耐热DNA聚合酶(Taq酶)将dNTP中的脱氧单核甘酸加到引物3’-OH末端,并以此为起点,沿着模板以5’→3’方向延伸,合成一条新的互补链。新合成的DNA链的起点是由加入的引物在模板DNA链两端的退火位点决定的。由于PCR反应中,双链DNA高温变性成单链,引物与模板单链DNA低温退火(配对),适温下引物延伸3个步骤反复循环,每一循环所形成的DNA分子均能成为下一循环的模板,所以PCR的特定目的DNA产物以指数方式递增,在数小时内,经过30个循环后,理论上可使DNA扩增至10亿(230=109)倍。并且原来对单拷贝基因进行探测和分析时需用10μg基因组DNA,现在可以减少到ng水平。样品DNA预变性双链DNA解链,94℃5min引物与单链模板DNA结合30-60℃,30秒-双链DNA解链聚合酶合成新链94℃、40秒65-75℃、2min延伸反应完成实验四PCR产物的纯化实验五DNA的酶切反应基因工程技术必不可少的工具酶之一,是限制性内切酶,它能识别和切割双链DNA10
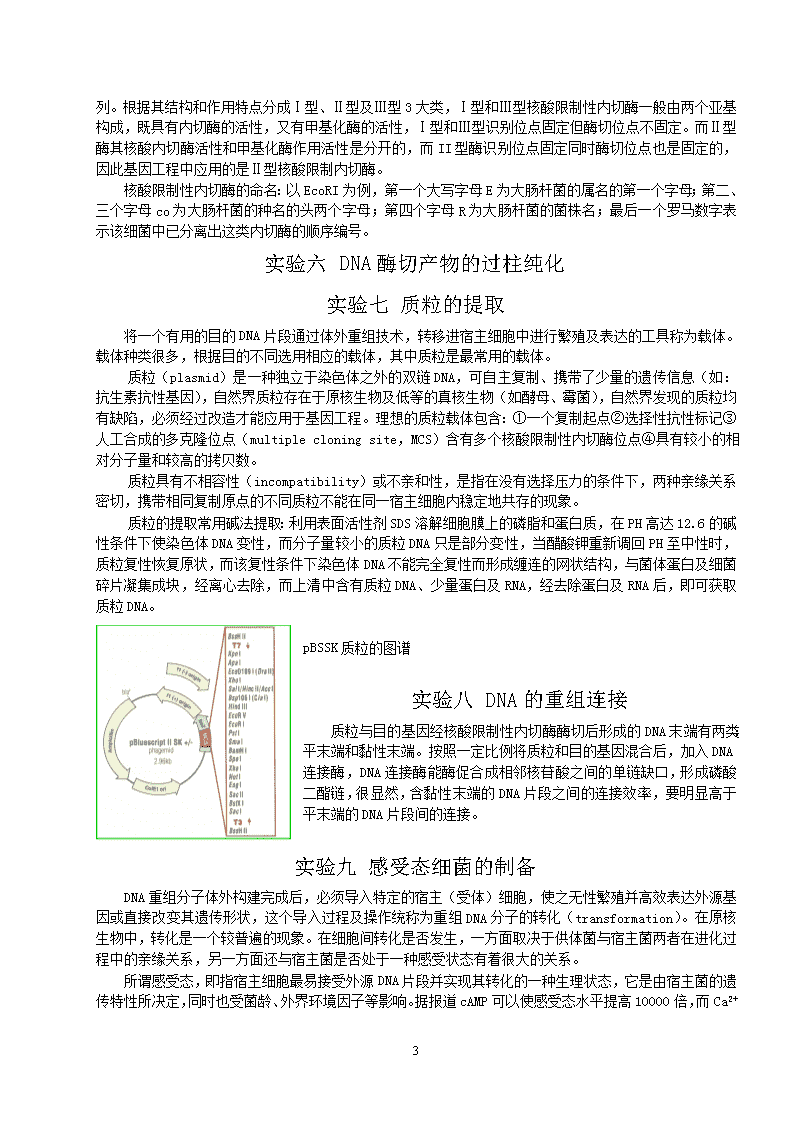
分子内特殊碱基序列。根据其结构和作用特点分成Ⅰ型、Ⅱ型及Ⅲ型3大类,Ⅰ型和Ⅲ型核酸限制性内切酶一般由两个亚基构成,既具有内切酶的活性,又有甲基化酶的活性,Ⅰ型和Ⅲ型识别位点固定但酶切位点不固定。而Ⅱ型酶其核酸内切酶活性和甲基化酶作用活性是分开的,而II型酶识别位点固定同时酶切位点也是固定的,因此基因工程中应用的是Ⅱ型核酸限制内切酶。核酸限制性内切酶的命名:以EcoRI为例,第一个大写字母E为大肠杆菌的属名的第一个字母;第二、三个字母co为大肠杆菌的种名的头两个字母;第四个字母R为大肠杆菌的菌株名;最后一个罗马数字表示该细菌中已分离出这类内切酶的顺序编号。实验六DNA酶切产物的过柱纯化实验七质粒的提取将一个有用的目的DNA片段通过体外重组技术,转移进宿主细胞中进行繁殖及表达的工具称为载体。载体种类很多,根据目的不同选用相应的载体,其中质粒是最常用的载体。质粒(plasmid)是一种独立于染色体之外的双链DNA,可自主复制、携带了少量的遗传信息(如:抗生素抗性基因),自然界质粒存在于原核生物及低等的真核生物(如酵母、霉菌),自然界发现的质粒均有缺陷,必须经过改造才能应用于基因工程。理想的质粒载体包含:①一个复制起点②选择性抗性标记③人工合成的多克隆位点(multiplecloningsite,MCS)含有多个核酸限制性内切酶位点④具有较小的相对分子量和较高的拷贝数。质粒具有不相容性(incompatibility)或不亲和性,是指在没有选择压力的条件下,两种亲缘关系密切,携带相同复制原点的不同质粒不能在同一宿主细胞内稳定地共存的现象。质粒的提取常用碱法提取:利用表面活性剂SDS溶解细胞膜上的磷脂和蛋白质,在PH高达12.6的碱性条件下使染色体DNA变性,而分子量较小的质粒DNA只是部分变性,当醋酸钾重新调回PH至中性时,质粒复性恢复原状,而该复性条件下染色体DNA不能完全复性而形成缠连的网状结构,与菌体蛋白及细菌碎片凝集成块,经离心去除,而上清中含有质粒DNA、少量蛋白及RNA,经去除蛋白及RNA后,即可获取质粒DNA。pBSSK质粒的图谱实验八DNA的重组连接质粒与目的基因经核酸限制性内切酶酶切后形成的DNA末端有两类:平末端和黏性末端。按照一定比例将质粒和目的基因混合后,加入DNA连接酶,DNA连接酶能酶促合成相邻核苷酸之间的单链缺口,形成磷酸二酯链,很显然,含黏性末端的DNA片段之间的连接效率,要明显高于平末端的DNA片段间的连接。实验九感受态细菌的制备DNA重组分子体外构建完成后,必须导入特定的宿主(受体)细胞,使之无性繁殖并高效表达外源基因或直接改变其遗传形状,这个导入过程及操作统称为重组DNA分子的转化(transformation)。在原核生物中,转化是一个较普遍的现象。在细胞间转化是否发生,一方面取决于供体菌与宿主菌两者在进化过程中的亲缘关系,另一方面还与宿主菌是否处于一种感受状态有着很大的关系。所谓感受态,即指宿主细胞最易接受外源DNA片段并实现其转化的一种生理状态,它是由宿主菌的遗传特性所决定,同时也受菌龄、外界环境因子等影响。据报道cAMP可以使感受态水平提高10000倍,而Ca2+10
也可大大促进转化作用。细胞的感受态一般出现在指数期,新鲜柔嫩的细胞是制备感受态细胞和进行成功转化的关键。对于Ca2+诱导的完整细胞转化而言,菌龄、CaCl2处理时间、感受态细胞(competentcell)的保存期以及热击时间均是很重要的因素,其中感受态细胞通常在12~24h内转化效率最高,之后转化率急剧下降。因此在制备感受态细胞时,将细胞培养至OD600为0.4~0.6后放入冰浴中使其终止生长,然后将菌株置于低温与处理的低渗CaCl2溶液中,即造成细胞膨胀,使细胞通透性发生短暂性变化,从而极易与外源DNA相黏附并在细胞表面形成复合物。此时,将该体系转移到42℃下做短暂的热击(heatshock),外源DNA就可能被细胞吸收。进入宿主细胞的DNA分子通过复制、表达,实现遗传信息的转移,是宿主细胞出现新的遗传性状。将经过转化的细胞在筛选培养基里培养,即可筛选出转化子(transformant),即带有异源DNA分子的宿主细胞。实验十重组质粒DNA的转化外源DNA与载体分子的连接即为DNA重组技术,重组的DNA分子式在DNA连接酶的作用下,有Mg2+、ATP存在的连接缓冲系统中,将分别经限制性内切酶酶切的载体分子和外源DNA分子连接起来。将重组质粒导入感受态细胞中,将转化后的细胞在选择性培养基中培养,可以通过各种方法筛选出重组子,并可通过酶切电泳进行重组子的鉴定。三、主要仪器设备在基因工程实验中用到的仪器主要包括:旋涡混合器、离心机、电泳槽、烘箱、PCR仪、冰箱等。此外,实验中所用工具还包括常见实验工具如:离心管、移胶板、eppendorf管、移液器等。四、操作方法和实验步骤实验一含GFP基因质粒的提取实验步骤:1.取菌液0.5~1ml,12000r/min离心10秒钟,弃上清液2.在旋涡混合器上振荡混匀至沉淀彻底分散3.加入400uL含溶菌酶的GTE溶液(含葡萄糖、Tris-Hcl缓冲液和EDTA),在旋涡混合器上振荡混匀至沉淀彻底分散4.37度保温20分钟。5.加酚-氯仿-异戊醇400ul,涡旋混匀,6.12000r/min离心5min,将上清液移至新的离心管中。7.向上清液中加入1/10体积(约40uL)的3mol/l醋酸钠(pH5.2),混匀,加入1mL的乙醇(约2倍体积的无水乙醇),混匀,-20度静止30min沉淀DNA,取出,12000r/min离心10min。8.小心弃上清液,用1mL的70%乙醇洗涤沉淀,12000r/min离心5min,弃上清,放入55度烘箱中3分钟,除去乙醇9.用50uL含RNaseA的TE溶解,37度温浴30min,除去RNA10.取5uL样品进行电泳。样品贮存再4度冰箱中。实验二琼脂糖DNA的电泳实验操作:1.称取0.7克的琼脂糖,加入100ml0.5×TBE,在微波炉上加热,使琼脂糖熔解2.等凝胶温度降至大约55℃以下时,加入5µl的0.5mg/L溴化乙锭(EB)。3.将移胶板放入胶室中,并将梳子垂直安插在移胶板上方,将凝胶倒入胶室中。10
4.等凝胶完全凝固后(约30分钟),将梳子轻轻拔出。5.将凝胶体放入加有0.5×TBE电泳缓冲液的电泳槽中,并且使电泳缓冲液高出凝胶。6.取2µl6×上样缓冲液与5µlPCR产物混匀后,加到凝胶孔中,7.加入如3µl的l/HindIIImarker*。8.盖好电泳槽盖子,选择适当的电泳电压(100V)以及电泳方向,开始电泳。9.前面色素接近胶的先端,切断电流,停止电泳,打开槽盖。10.取出样品,在紫外灯下观察,摄像。实验三PCR扩增目的基因实验操作1.取一个0.2mleppendorf管,添加以下各种成分反应液ddH2O36.5µl模板DNA5µl(100-200ng)*1上游引物(10µmol/L)1µl下游引物(10µmol/L)1µldNTPmixture(10mmol/L)1µl(各20nmol)10倍×缓冲液+MgCl25µlTaq酶0.5µl(2.5U)总体积50µl2.稍作离心,将反应管放入PCR仪中。3.设定反应程序:94℃条件下使模板DNA预变性5min。变性94℃30秒退火58℃30秒30cycles延伸72℃1.0min最后在72℃条件进行延伸7-10min。4℃保存4.取5µl反应液用0.7%琼脂糖凝胶进行电泳,鉴定PCR产物是否存在以及大小。实验四PCR产物的纯化实验操作1.电泳确认后将PCR产物移至1.5ml新的eppendorf管中。2.加入200µlTE缓冲液,加入300µl酚/氯仿/异戊醇,混匀,室温,12000rpm离心5分钟。*3.取上层水液添加1/10体积(约30µl)的3mol/L的NaAc(pH5.2)和2.5倍体积(约0.75ml)的冰冷无水乙醇。4.-20℃冰箱中放置30min以上。5.4℃、12000rpm离心5min。6.弃上清,添加1ml的冰冷70%乙醇。4℃、12000rpm离心5min。7.弃去上清,室温干燥,30µlTE液溶解沉淀。实验五DNA的酶切反应实验步骤:1.在0.5mleppendorf管中加入下列成分,(定容40µl)A10
ddH2O2µlPCR产物30µl(5µg)10×缓冲液4µlXhoⅠ2µlBamHI2µl总体积40µlBddH2O2µlpET质粒30µl10×缓冲液4µlXhoⅠ2µlBamHI2µl总体积40µl2.混匀,少许离心一下。3.在37℃条件下反应2-4小时以上,或酶切过夜。4.取5µl酶切质粒进行凝胶电泳,预检。实验六DNA酶切产物的过柱纯化1.反应管中加BufferB3250µl,轻弹混合均匀2.装柱,将pET的双酶切产物混合液加在柱子白色填料区,静置1分钟,8000rpm离心1分钟并弃去上层废液。3.分别加入500µlWashsolution,9000rpm离心30s弃去废液。4.重复步骤3,弃去废液后再在9000rpm条件下空离1分钟。5.分别将两个柱子转入新管,在柱中心加20µlElution,静置1分钟,离心1分钟。收集产物。实验七质粒的提取实验步骤1.取1.5ml培养液在4℃、12000rpm离心2min,弃去上清液,收集菌体。2.加入100µl的溶液I,使菌体充分悬浮,室温条件下静置5min。3.加入200µl新配置的溶液II,温和上下颠倒2-3回,冰浴条件下静置5min。4.加入150µl的溶液III,上下颠倒混匀数次,冰浴条件下静置5min。5.4℃条件下12000rpm离心10min。6.取上清,加入等体积(400µl)的酚/氯仿/异戊醇,充分混匀,室温条件下12000rpm离心5min。7.吸取上层水相,加入1/10体积量(约40µl)3mol/L的pH5.2NaAc,再加入2.5倍体积(约1ml)的预冷的无水乙醇。8.放入-20℃冰箱中静止30min,然后在4℃条件下12000rpm离心15min。9.弃去上清液,加入1ml预冷的70%乙醇,在4℃条件下12000rpm离心5min,洗涤沉淀。10.弃上清液,沉淀物自然干燥后,加入40µlTE缓冲液溶解沉淀(pET质粒用25TE溶解)。11.加入2µl的10mg/ml的RNaseA酶,37℃保温20-30min。12.取5µl的质粒电泳。实验八DNA的重组连接10
实验步骤:1.在离心管中加入以下样品:PCR产物6µlpET/酶切产物2µl10×连接缓冲液1µlT4DNA连接酶1µl2.混匀后,离心将液体全部甩到管底。3.16℃条件下保温过夜。4.-20℃备用实验九感受态细菌的制备实验步骤:1.接种BL21于LB培养基中37℃振荡培养过夜。2.接100µl培养液于5ml的LB液体培养基中,37℃条件下振荡培养2-2.5小时左右,OD600达0.4。3.将1.5ml培养液移至eppendorf管中、冰浴20min。4.4℃条件下,4000rpm离心5min,弃去上清液。5.添加0.75ml预冷的CaCl2溶液,用枪轻轻吹打。6.冰浴30分钟后,4℃条件下,4000rpm离心5min,弃去上清液。7.细胞沉淀用100µl预冷的CaCl2溶液悬浮。8.冰浴放置,备用。实验十重组质粒DNA的转化1.取一管100µl的感受态细胞,加入质粒与目的基因的连接产物5µl,轻轻用枪吹打均匀,冰浴20min2.42℃、保温90秒钟,迅速放入冰中,冰浴5min。3.添加1ml的LB培养基。4.37℃振荡培养1小时。5.3000rpm离心5分钟,弃去1ml上清,余下0.1ml用枪吹匀,吸至含有抗生素的LB培养基平板上,均匀涂布。6.37℃倒置培养8-12小时。五、实验结果与分析图110
上图为GFP双酶切回收的电泳结果,从图中可以看出,在实验一:含GFP基因质粒的提取中,还是比较成功的,即获取了目标基因组的DNA。图2上图为PCR扩增后的电泳结果(左五)从图中可以看出,在此次电泳中,我的PCR产物并没有很成功地产生。由于在实验一中的结果并没有出现问题,因此我分析实验三的电泳结果之所以会失败,应该是在实验二或是实验三的过程中出现了操作失误或是其他不可控因素。在实验三的操作步骤中,主要部分为取一个0.2mleppendorf管,添加各种成分反应液。由于各种反应液要添加的量都比较小,而且添加顺序比较固定,不能出现乱序添加的情况,这些都是可能造成实验失败的因素。但经过回忆,我在添加各反应液的过程中使用的是设定好吸取容量的移液器,也没有出现容量错误、吸取排出不到位等现象。由于是排队添加,顺序与周围同学进行添加的顺序也是一致的,因此我分析在添加反应液这一步没有出现失误。根据实验步骤,在添加反应液过后应该稍作离心,再放入PCR仪中进行PCR扩增。我在添加反应液之后,因为想使各种反应液更好融合,在离心之前将eppendorf管在旋涡反应器上进行了时长约15秒的涡旋。我猜想实验结果的失败很有可能与这次涡旋有关。在涡旋的过程,振荡太过激烈,破坏了原有的基因组DNA,因此在之后的PCR扩增中没有出现产物。鉴于这一步骤的实验失败,为了顺利进行接下来的实验,我吸取了老师提供的PCR扩增产物以完成随后的实验。10
图3上图为荧光蛋白的电泳结果(左三),从图中可以看到,虽然显示不是特别明显,但是基本还算比较成功,可以作为荧光蛋白的培养菌落。图410
上图为实验最后的荧光蛋白结果,从图中可以看到,荧光蛋白成功培养了出来,亮点清晰,但是数量较少,密度较小。总体来说算是完成了整个基因工程实验的实验要求,但是由于某些因素造成了菌落生长的不均匀与小数量。我分析出现这种情况的原因大概跟我在将细胞铺匀到平板上是出现了操作失误。在将反应管中的细胞液吸至平板的过程中,我错误地将一半左右的细胞液铺在了没有营养的平板盖上面。虽然在金老师的帮助下将那一部分细胞转移到了正确的位置,但是有可能在这个过程中使细胞收到了污染,也就造成了一半容量左右的细胞失去了生长的能力。六、讨论、心得毫不夸张地说,此次基因工程实验是迄今为止我做过的耗时最长、所用仪器设备最高端精良、涉及专业知识最有深度的一次。在此次实验中,我更加认识到了实验本身所能够具有的魅力以及生物的奇妙,总体来说,这还是一次非常有意义、值得纪念的经历。同时我不得不承认,在实验进行的过程中,我对老师所讲解的原理并不是非常理解。作为一名文科生,接触生物这一学科的时间大致只有从初一到高二断断续续的三年。初中所教的生物知识比较浅显,而到了高中,由于我选择了文科方向,高二之后就没有再接触过生物实验,对于基本的生物知识和实验规范也没有很深刻的理解和认识。虽然并不能完全理解和掌握基因工程实验中涉及到的全部专业知识和原理,但是在实验过程中我尽力跟紧老师上课的节奏,并在动手实践的过程中对每一步的原理进行思考并加深印象。从分不清不同移液器的作用与容量到熟练操作三种移液器并了解到吸取微量液体的注意事项,从对电泳的印象只是化学课本上的一张图到真正动手去体验电泳的过程,从丝毫不了解细胞的生长与反应到最终成功收获荧光蛋白…这次生物实验带给我的不仅仅是各种与生物有关的知识和实验操作的规范与常识,还有对微观生物这个崭新的世界的好奇与热情,更是一种不断学习不断接触新事物的体验。从这个意义上来说,一次生物实验所代表的不仅仅是实验,也代表着生物这门学科的精髓与核心魅力,代表着不断尝试不断进取的激情与活力。除了实验本身的过程带给人的感受,在实验中悉心知道我们的赵老师和金老师也给我们留下了很深的印象。第一次进行这种专业性的实验,同学们普遍都有一些慌张和经验不足。两位指导老师在两天的时间里一直全力对我们进行帮助。金老师为我们提前做好了很多实验中会用到的东西例如凝胶等,也在包括我在内的几位同学获取PCR产物失败后为我们提供产物,以确保我们可以继续进行实验。两位老师为这个实验付出了很多时间和精力,非常感谢老师们的帮助。虽然此次实验已经结束了,但是这段经历所带给我的感受会一直保存下去,希望今后还可以有机会走进实验室,感受生物的魅力所在。10