


- 251.50 KB
- 11页


- 1、本文档共5页,可阅读全部内容。
- 2、本文档内容版权归属内容提供方,所产生的收益全部归内容提供方所有。如果您对本文有版权争议,可选择认领,认领后既往收益都归您。
- 3、本文档由用户上传,本站不保证质量和数量令人满意,可能有诸多瑕疵,付费之前,请仔细先通过免费阅读内容等途径辨别内容交易风险。如存在严重挂羊头卖狗肉之情形,可联系本站下载客服投诉处理。
- 文档侵权举报电话:19940600175。
暴露在一氧化碳的研究课题发现血碳酸正常可改善常规氧治疗时的脑氧交付JoshuaRucker,MDJanetTesler,MScLudwikFedorko,MD,PhDAkinoriTakeuchi,MD,PhDLucianaMascia,MDAlexVesely,MScSashaKobrossi,BScArthurS.Slutsky,MDGeorgeVolgyesi,PEngSteveIscoe,PhDJosephA.Fisher,MDFromtheDepartmentofAnesthesia,UniversityHealthNetwork,UniversityofToronto,Toronto,Ontario,Canada(Rucker,Tesler,Fedorko,Mascia,Vesely,Kobrossi,Volgyesi,Fisher);theDepart-mentofAnesthesiaandResuscitology,NagoyaCityUniversityMedicalSchool,Nagoya,Japan(Takeuchi);theDepartmentofCriticalCareMedicineandDepartmentofMedicine,St.Michael’sHospital,Toronto,Ontario,Canada(Slutsky);andtheDepartmentofPhysiology,QueensUniversity,Kingston,Ontario,Canada(Iscoe).研究目的:判断在使用高氧治疗一氧化碳中毒期间维持血碳酸正常是否能改善脑氧交付。方法:本实验使用随机的,单盲的,交叉的设计。我们会把14个人体研究对象暴露在一氧化碳中,直到他们的碳氧血红蛋白水平到达10%-12%。然后每位研究对象进行60min血碳酸正常或没有血碳酸正常的高氧治疗。把通过至少24小时治疗后得到恢复的研究对象再次暴露在一氧化碳中,并进行替代治疗。脑氧交付的相关变化可以通过经颅多普勒超声计算测量血氧含量产物和中脑动脉流速(脑血流指数)得出。结果:与标准氧治疗(P<0.05;60min内95%的置信区间在2.8%-16.7%)方法相比,在高氧治疗中维持血碳酸正常由于防止低血碳酸引起大脑血管收缩可使脑氧交付显著提高,并且由于提高每分钟通气量可加快一氧化碳清除速度。总结:如我们的研究对象,属于重度中毒患者,在一氧化碳中毒进行高氧治疗初期开始维持血碳酸正常会提高氧气传输到大脑。需要临床研究才能判断常规治疗这种变化的结果的效果。介绍一氧化碳吸入过多是工业世界致命中毒的主要原因。在美国,每年有40000个由于一氧化碳中毒而引发的急诊病例,其中大约有3500-4000个死亡病例。一氧化碳中毒的重要原理的血红蛋白中的氧气被一氧化碳置换出来从而导致组织缺氧。大脑和心脏通常是受影响最严重的器官,因为它们对组织缺氧最敏感。用高氧进行一氧化碳中毒急救治疗的理由是,第一,通过提高血氧含量快速增加组织氧合;第二,通过促进一氧化碳清除可缩短血液携氧能力的恢复时间。然而,脑氧交付是血氧含量和脑氧流量(CBF)的一项功能。在健康调查中人们很早发现CBF可以降低对高压氧的反应。其中一个关于CBF提出的高氧联结机制是通过氧气的作用作为呼吸兴奋剂,从而导致Paco2的下降。CBF对Paco2的下降十分敏感,在Paco2低于40mmHg(5.3kPa)时每mmHg(0.133kPa)
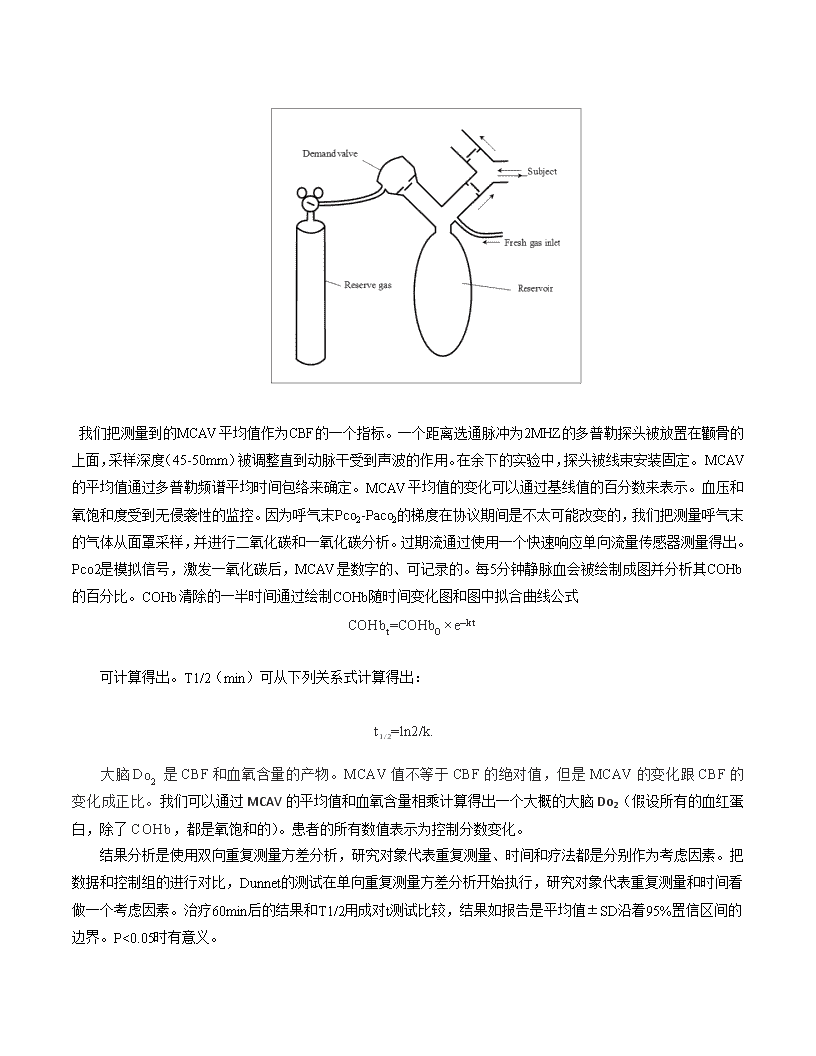
大约下降2%。高氧治疗伴随的低碳酸血症而导致的任何脑灌注下降都会抵消大脑输氧量的预期增加。本次研究的目的是比较计算在一氧化碳暴露中研究对象进行高氧治疗的过程中是否采取措施预防低碳酸血症的脑氧交付(Do2)。材料和方法在收到来自机构人类学科审查委员会人体试验的批准后,我们得到了年龄在18-47岁、已签署知情同意书的9名男性和9名女性作为研究对象。所有的研究对象均没有吸烟史、没有服用药物、体重正常。在方案执行前,所有研究对象先在医院的临床实验室进行静脉血血红蛋白浓度测定和肺功能测试。为了把黄酮素对通气的影响降到最低,女志愿者会在经期前两周进行研究。志愿者会在不同的日期分为两阶段进行测试。当基线测量确定后,把在控制周期的研究对象暴露在一氧化碳中,然后选择两个随机治疗方案中的一个。治疗由呼吸有或没有增加以维持血碳酸正常的二氧化碳的富氧气体组成。治疗顺序是通过随机挑选贴有记号的外包装来确定。呼吸回路是平衡的,因此研究对象无法辨别实验阶段或治疗类型。研究对象舒适的坐着,前臂静脉插入留置导管。他们用带有硅胶密封条的全面罩通过回路进行呼吸。初始激发气体是空气,研究对象适应面罩和回路(10-20min)。控制措施开始后(10min),研究对象被暴露在1,000±200ppm一氧化碳的空气中直到他们的碳氧血红蛋白(COHb)水平达到10%-12%(30-60min)。每5分钟对静脉血样本进行一次碳氧血红蛋白分析。在一氧化碳暴露后,研究对象用100%氧气(poikilocapnichyperoxia)或用96%-100%的氧平衡二氧化碳进行治疗,随着每分钟通气量的增加,二氧化碳用于维持血碳酸正常(血碳酸正常的高氧)。在之前已经描述了回路,包括一个非重复呼吸阀,一个连接氧气囊并直接导入氧气的入气口(图1)。如果每分通气量超过氧气流量,增加气体,储备气体从压缩气缸调节流量得以利用。对于poikilocapnic治疗,储备气体由氧气组成;对于normocapnic治疗,储备气体由大约6%的二氧化碳平衡氧组成。在一定程度上,每分钟通气量超过静止水平,储备气体按正比例增加提供二氧化碳作为激发气体。二氧化碳的浓度大约为6%,这样的Pco2是为了防止二氧化碳清除和呼气末Pco2的改变。通过调节静止时氧气流量略低于静止时每分钟通气量,研究对象可在静止时激发储备气体,这样通过开启流量调节器并未留意到通气量增加。每分钟通气量高于静止水平的任何增加都会增加一氧化碳而非二氧化碳的血液到肺泡的分压梯度,因此在防止低血碳酸的同时可提高一氧化碳的清除。图1.回路的原理图.新鲜气体通过新鲜气体进气口输送并充满气体储备囊。若储备囊为空,打开需求阀,患者呼吸储备气体(100%O2或6%CO2平衡O2)。
我们把测量到的MCAV平均值作为CBF的一个指标。一个距离选通脉冲为2MHZ的多普勒探头被放置在颧骨的上面,采样深度(45-50mm)被调整直到动脉干受到声波的作用。在余下的实验中,探头被线束安装固定。MCAV的平均值通过多普勒频谱平均时间包络来确定。MCAV平均值的变化可以通过基线值的百分数来表示。血压和氧饱和度受到无侵袭性的监控。因为呼气末Pco2-Paco2的梯度在协议期间是不太可能改变的,我们把测量呼气末的气体从面罩采样,并进行二氧化碳和一氧化碳分析。过期流通过使用一个快速响应单向流量传感器测量得出。Pco2是模拟信号,激发一氧化碳后,MCAV是数字的、可记录的。每5分钟静脉血会被绘制成图并分析其COHb的百分比。COHb清除的一半时间通过绘制COHb随时间变化图和图中拟合曲线公式COHbt=COHb0×e–kt可计算得出。T1/2(min)可从下列关系式计算得出:t1/2=ln2/k.大脑DO2是CBF和血氧含量的产物。MCAV值不等于CBF的绝对值,但是MCAV的变化跟CBF的变化成正比。我们可以通过MCAV的平均值和血氧含量相乘计算得出一个大概的大脑Do2(假设所有的血红蛋白,除了COHb,都是氧饱和的)。患者的所有数值表示为控制分数变化。结果分析是使用双向重复测量方差分析,研究对象代表重复测量、时间和疗法都是分别作为考虑因素。把数据和控制组的进行对比,Dunnet的测试在单向重复测量方差分析开始执行,研究对象代表重复测量和时间看做一个考虑因素。治疗60min后的结果和T1/2用成对t测试比较,结果如报告是平均值±SD沿着95%置信区间的边界。P<0.05时有意义。
结果其中1名男性和3名女性研究对象是排除在外的,因为我们在初始控制组没有得到满意的MCAV信号。其余研究对象的人体测量和肺功能数据仍然会保存在表中。表.本研究中所有研究对象的体位数据和控制值表poikilocapnichyperoxia随着持续治疗每分钟通气量不断增加和呼气末Pco2不断下降(P<0.01,如图2),这表明对高氧有反应的呼吸道仍然存在高达12%的COHb。与之相反,normocapnichyperoxic治疗在Pco2(P=0.4)不变的情况下使每分钟通气量(P<0.001)更大的提高,这里通气量的增加是与12±9分钟(95%置信区间7-17)或一氧化碳清除的T1/2降低到21%有关(45±8随着57±11min;P<0.05)。图2在Poikilocapnic(●)和normocapnic(〇)治疗中,每分钟通气量(VE)和呼气末PCO2随时间变化图,*表示治疗在2-wayRM-ANOVAs基础上的显著性差异
在控制条件下暴露在一氧化碳中可提高MCAV到12.8±9.9%(95%置信区间9.1-16.4;P<0.01)。一旦过度通气引起的氧反应成立,MCAV在poikilocapnichyperoxia中比血碳酸正常下降更多(P<0.001;图3)。在研究之末,poikilocapnichyperoxia时MCAV值是9.4±13.6%(95%置信区间-2.2到-16.5;P<0.05),与血碳酸正常相比的低。受到CBF和COHb影响的脑氧交付在血碳酸正常氧治疗中明显大于poikilocapnichyperoxia(9.7±13.3%;95%置信区间2.8-16.7;P<0.05)。我们可以观察到在poikilocapnichyperoxia时,大脑的DO2在不同研究对象的影响是变化的。尽管某些研究对象通过高氧治疗脑氧交付没有明显的下降,而其余的研究对象表示有显著的减少。其中的3名研究对象脑氧交付下降大于20%(5号、12号、13号)图3.在poikilocapnic(●)和normocapnic(〇)氧治疗CO中MCAV和大脑DO2随时间的变化。*表示在控制下的显著性差异。†表示双向重复测量方差分析的显著性差异。
讨论研究表明,使用100%氧治疗一氧化碳中毒患者会导致CBF下降,完全抵消脑氧交付中增加血氧含量的效果。而且,维持血碳酸正常不仅防止CBF和脑氧交付的减少,而且能促进清除一氧化碳。结果符合Kreck等人的观察,Kreck等人证明了保持高氧正常血碳酸,在一氧化碳中毒患者进行机械通气,可改善全身氧运输和一氧化碳清除。然而,他们既没有评估血碳酸正常的独立作用,也没有对脑氧交付进行明确测量。机械给氧的刺激作用直到今天也没有得到大家的普遍认同,尽管在一个世纪以前其就在研究和测试中被反复描述。Lambertsen等人指出,CBF的减少与高氧有关,并且往往伴随着Paco2的下降。我们的工作延伸到观察高氧,低血碳酸和CBF的降低的关系以证明这个机制保留存在5%-12%COHb。从20世纪20年代到二战结束,二氧化碳结合氧气治
疗一氧化碳中毒这样的方法一直被沿用。这个方法在1922年由Henderson和Haggard报道。Haggard曾报道过严重一氧化碳中毒患者使用卡波金法治疗后快速复苏,即一定比例的二氧化碳和氧气的混合气体。他们归结出提高卡波金法效果是往氧气中添加了二氧化碳的通气刺激。我们的研究建议他们的病人也得益于脑氧交付的初始冲击效果。然而,根据每分钟通气量,利用卡波金法治疗会导致低血碳酸或高血碳酸。与之相反,我们的研究维持独立于每分钟通气量且血碳酸正常的循环。最近,Sasano等人对我们的通过简单改进的、普遍的自动充气包进行了简单描述。血液中存在的一氧化碳会减少携氧能力,满足大脑对灌注的依赖以满足它对氧气的需求。在我们的研究中,一氧化碳暴露末端,携氧能力下降10%(由于碳氧血红蛋白的作用结果)会抵消CBF增加的13%,相当于对脑氧交付没有任何作用。两种疗法中,含氧量提高不明显,但使用高氧疗法后,CBF在某些研究对象中的下降十分明显。在那些Pco2下降最多的研究对象中,CBF和脑氧交付在控制下分别下降33%和26%。CBF的下降与Watson等人在报告中的是相似的,Watson观察到没有暴露在一氧化碳中而呼吸100%氧气的研究对象的CBF减少了31%。甚至在没有一氧化碳中毒的病人中,CBF减少的等级也与症状性脑缺血有关,进一步加强了在没有维持血碳酸正常的氧治疗会导致副作用,甚至有不良后果。一氧化碳中毒同样导致血红蛋白对氧的亲和力的增加,削弱氧对组织的卸载能力。如果患者对高氧治疗出现低血碳酸症状,由于波尔效应,氧合血红蛋白游离曲线会向左偏离。一般来说,维持血碳酸正常会通过防止CBF的降低而改善脑氧交付,通过最小化的氧合血红蛋白解离曲线的左支会转变,会促进一氧化碳清除。我们的研究中有很多限制因素。第一,因为CBF不能直接测量,我们使用MCAV作为CBF的指标。MCAV方法假设大脑中动脉的一个恒定值,Pco2的有限范围根据研究对象来确定。人类在大脑中动脉直径上对一氧化碳的作用知之甚少,但有一个研究显示一氧化碳并不能扩张延伸活体兔的脑血管。在任何情况下一氧化碳扩张我们研究对象的大脑中动脉,结果导致MCAV的下降会被解读成CBF似是而非的下降;事实并非如此。第二个限制因素涉及到我们把研究结果外推到患者。我们的研究对象的碳氧血红蛋白水平跟一氧化碳中毒患者相比相对较低。然而,我们预计二氧化碳对脑血管的反应性持续在更高的碳氧血红蛋白水平并存在脑缺氧。低血碳酸症容易的废除高浓度一氧化碳的轻度直接血管扩张的作用由于低血碳酸存在高浓度的如此强大的脑血管扩张剂如氟烷和异氟醚使CBF降低。而且,适度的低血碳酸(27毫米汞柱的Paco2)能过渡甚至更严重的低氧血症的脑血管扩张的影响(40毫米汞柱的Paco2)。第三个限制因素是我们的研究集中在脑氧交付的变化,在氧气治疗时
排除其它机制治疗一氧化碳中毒。这些包括一氧化氮介导组织损伤,加速细胞凋亡和干扰细胞内养的运输。在一氧化碳暴露的未知程度和持续时间之后提高脑氧交付对大脑造成伤害程度的影响对一个病人来说是难以预计的。然而,因为大多数一氧化碳中毒直接作用是依赖或加重组织缺氧,这就有足够的理由建议在治疗早期就最优化脑氧交付在治疗一氧化碳中毒上仍然是一个重要目标。维持血碳酸正常意味着提高脑氧交付。例如,这或许也影响大脑区域血流分布,抑制细胞内氧气运输和使用,并且造成一定程度的再灌注损伤。在严重中毒的患者中维持血碳酸正常的净效应是难以预计和需要更深的研究,特别是那些关注结果的。缺乏措施维持血碳酸正常,一氧化碳中毒患者可以用氧气治疗吗?尽管我们的结果揭露我们一氧化碳暴露的研究对象在脑氧交付中氧气的干扰下降,我们不能确定病人的受影响程度或组织的损伤程度或结果。这些问题在实验强制发生变化前会被解决。综上所述,我们可以证明轻度一氧化碳中毒研究对象用高氧治疗会降低大脑血流量和大脑氧运输。维持血碳酸正常能防止CBF的降低和促进一氧化碳的清除,改善脑氧交付。参考文献1.FisherJA,SommerLZ,RuckerJ,etal.Isocapnichyperpneaacceleratescarbonmonoxideelimination.AmJRespirCritCareMed.1999;159:1289-1292.2.HampsonNB.EmergencydepartmentvisitsforcarbonmonoxidepoisoninginthePacificNorthwest.JEmergMed.1998;16:695-698.3.CobbN,EtzelRA.Unintentionalcarbonmonoxide-relateddeathsintheUnitedStates,1979through1988.JAMA.1991;266:659-663.4.WeaverLK.Carbonmonoxidepoisoning.CritCareClin.1999;15:297-317.5.OlsonKR.Carbonmonoxidepoisoning:mechanisms,presentation,andcontrover-siesinmanagement.JEmergMed.1984;1:233-243.6.KetySS,SchmidtCF.Theeffectsofalteredarterialtensionsofcarbondioxideandoxygenoncerebralbloodflowandcerebraloxygenconsumptionofnormalyoungmen.JClinInvest.1948;27:484-492.7.DautrebandeL,HaldaneJS.Theeffectsofrespirationofoxygenonbreathingandcirculation.JPhysiol.1921;55:296-299.8.BeckerH,PoloO,McNamaraSGetal.Ventilatoryresponsetoisocapnichyper-oxia.JApplPhysiol.1995;78:696-701.9.FortuneJB,BockD,KupinskiAM,etal.Humancerebrovascularresponsetooxy-genandcarbondioxideasdeterminedbyinternalcarotidarteryduplexscanning.JTrauma.1992;32:618-627.10.BrianJEJr.Carbondioxideandthecerebralcirculation.Anesthesiology.1998;88:1365-1386.11.DuttonK,BlanksbyBA,MortonAR.CO2sensitivitychangesduringthemenstrualcycle.JApplPhysiol.1989;67:517-522.
12.SommerLZ,IscoeS,RobicsekA,etal.Asimplebreathingcircuitminimizingchangesinalveolarventilationduringhyperpnoea.EurRespirJ.1998;12:698-701.13.BishopCCR,PowellS,RuttD,etal.TranscranialDopplermeasurementofmiddlecerebralarterybloodflowvelocity:avalidationstudy.Stroke.1986;17:913-915.14.MarkwalderTM,GrolimundP,SeilerRW,etal.Dependencyofbloodflowvelocityinthemiddlecerebralarteryonend-tidalcarbondioxidepartialpressure–atranscra-nialultrasoundDopplerstudy.JCerebBloodFlowMetab.1984;4:368-372.15.KreckTC,ShadeED,LammWJ,etal.Isocapnichyperventilationincreasescarbonmonoxideeliminationandoxygendelivery.AmJRespirCritCareMed.2001;163:458-462.16.YamadaM.MethodischeUntersuchungenüberdasHaldane-HendersonscheVerfahrenderBestimmungderalveolärenCO2SpannungundüberdenEinfluβvonSauerstoffaufdieErregbarkeitdesAtemzentrums.BiochemZtschr.1918;89:27-47.17.ComroeJH,DrippsRD.ThePhysiologicalBasisforOxygenTherapy.1sted.Springfield,IL:CharlesC.Thomas;1950.18.LambertsenCJ,StroudMW,GouldRH,etal.Oxygentoxicity.Respiratoryre-sponsesofnormalmentoinhalationof6and100percentoxygenunder3.5atmo-spherespressure.JApplPhysiol.2001;5:487-494.19.DonaldKW,PatonWDM.Gasesadministeredinartificialrespiration:withparticu-larreferencetotheuseofcarbondioxide.BMJ.1955;313-318.20.HendersonY,HaggardHW.ThetreatmentofcarbonmonoxidasphyxiabymeansofoxygenandCO2inhalation.JAMA.1922;79:1137-1145.21.HoskinPJ,AbdelathO,PhillipsH,etal.Inspiredandexpiredgasconcentrationsinmanduringcarbogenbreathing.RadiotherOncol.1999;51:175-177.22.SasanoH,VeselyAE,IscoeS,etal.Asimpleapparatusforacceleratingrecoveryfrominhaledvolatileanesthetics.AnesthAnalg.2001;93:1188-1191.23.WatsonNA,BeardsSC,AltafN,etal.Theeffectofhyperoxiaoncerebralbloodflow:astudyinhealthyvolunteersusingmagneticresonancephase-contrastangiogra-phy.EurJAnaesthesiol.2000;17:152-159.24.StrandgaardS,OlesenJ,SkinhojE,etal.Autoregulationofbraincirculationinseverearterialhypertension.BMJ.1973;1:507-510.25.RoughtonFJW,DarlingRC.Theeffectofcarbonmonoxideontheoxyhemoglobindissociationcurve.AmJPhysiol.1944;141:17-31.26.HlastalaMP,McKennaHP,FranadaRL,etal.Influenceofcarbonmonoxideonhemoglobin-oxygenbinding.JApplPhysiol.1976;41:893-899.27.BrianJEJr.,HeistadDD,FaraciFM.Effectofcarbonmonoxideonrabbitcerebralarteries.Stroke.1994;25:639-643.
28.LefflerCW,NasjlettiA,YuC,etal.Carbonmonoxideandcerebralmicrovasculartoneinnewbornpigs.AmJPhysiol.1999;276:H1641-H1646.29.ChongKY,CraenRA,MurkinJM,etal.Rateofchangeofcerebralbloodflowvelocitywithhyperventilationduringanesthesiainhumans.CanJAnaesth.2000;47:125-130.30.YoungWL,ProhovnikI,OrnsteinE,etal.Cerebralbloodflowreactivitytochangesincarbondioxidecalculatedusingend-tidalversusarterialtensions.JCerebBloodFlowMetab.1991;11:1031-1035.31.ShapiroW,WassermanAJ,BakerJP,etal.Cerebrovascularresponsetoacutehypocapnicandeucapnichypoxiainnormalman.JClinInvest.1970;49:2362-2368.32.IschiropoulosH,BeersMF,OhnishiST,etal.Nitricoxideproductionandperivas-cularnitrationinbrainaftercarbonmonoxidepoisoningintherat.JClinInvest.1996;97:2260-2267.33.ThomSR.Carbonmonoxide-mediatedbrainlipidperoxidationintherat.JApplPhysiol.1990;68:997-1003.34.PiantadosiCA,ZhangJ,LevinED,etal.Apoptosisanddelayedneuronaldamageaftercarbonmonoxidepoisoningintherat.ExpNeurol.1997;147:103-114.35.TurcanuV,DhouibM,GendraultJL,etal.Carbonmonoxideinducesmurinethymo-cyteapoptosisbyafreeradical-mediatedmechanism.CellBiolToxicol.1998;14:47-54.36.CoburnRF.Thecarbonmonoxidebodystores.AnnNYAcadSci.1970;174:11-22.37.PiantadosiCA.Carbonmonoxide,oxygentransport,andoxygenmetabolism.JHyperbaricMed.1987;2:27-44